Proc. Natl. Acad. Sci. | AlphaFold3在预测配体诱导酶结构域运动中的偏差

Proc. Natl. Acad. Sci. | AlphaFold3在预测配体诱导酶结构域运动中的偏差

DrugAI
发布于 2026-03-30 18:42:32
发布于 2026-03-30 18:42:32
DRUGONE
蛋白质在执行功能时通常需要发生构象变化,尤其是在配体结合后产生的结构域运动对酶活性至关重要。研究人员系统评估了AlphaFold3在预测配体诱导的结构域运动方面的能力,发现尽管该模型在静态结构预测方面表现优异,但在涉及大尺度构象变化时存在明显偏差。通过构建包含已知开放态和闭合态结构的酶数据集,并比较有无配体条件下的预测结果,研究人员发现AlphaFold3往往倾向于预测接近训练集中常见构象的结构,而不能准确再现配体诱导的构象转换。该结果提示,在研究酶催化机制和配体调控时,需要谨慎解读AlphaFold3预测的构象变化。

蛋白质结构预测的突破性进展使得研究人员能够在没有实验结构的情况下获得高精度三维模型。AlphaFold系列模型在预测静态蛋白结构方面取得了显著成功,并逐渐扩展到多聚体和蛋白-配体复合物的预测。然而,许多酶在催化过程中会经历显著的结构域运动,例如开闭状态转换、铰链运动或诱导契合,这些动态变化对理解催化机制至关重要。
尽管AlphaFold3可以在输入中加入配体信息,从而预测复合物结构,但其训练主要基于已解析的静态结构数据,因此可能存在对常见构象的偏好,从而影响对配体诱导构象变化的预测。此前已有个别案例显示AlphaFold在预测构象变化时可能偏向稳定态,但尚缺乏系统性的评估。为此,研究人员建立了一个包含多种酶类的测试集合,对AlphaFold3在配体诱导结构域运动预测中的表现进行系统分析,以明确其优势和局限。
方法
研究人员收集了一组具有明确开放态和闭合态晶体结构的酶,并确保这些结构对应不同配体结合状态。对于每个体系,分别使用AlphaFold3在无配体和有配体条件下进行预测,并生成多个结构模型。随后通过计算结构域间距离、旋转角度以及整体构象差异,对预测结构与实验结构进行比较,并评估模型是否能够正确再现配体诱导的结构变化。此外,还分析了不同输入设置、模板使用情况以及配体信息对预测结果的影响,以确定导致偏差的主要因素。
结果
测试数据集与构象变化示例
研究人员构建的测试集包含多种具有显著结构域运动的酶,例如在配体结合后发生闭合的转移酶和脱氢酶。实验结构显示这些蛋白在无配体时处于开放状态,而在配体结合后形成紧密的闭合构象。对这些体系进行AlphaFold3预测后发现,模型通常生成接近其中一种已知构象的结构,而不是根据输入配体准确切换到另一种状态。
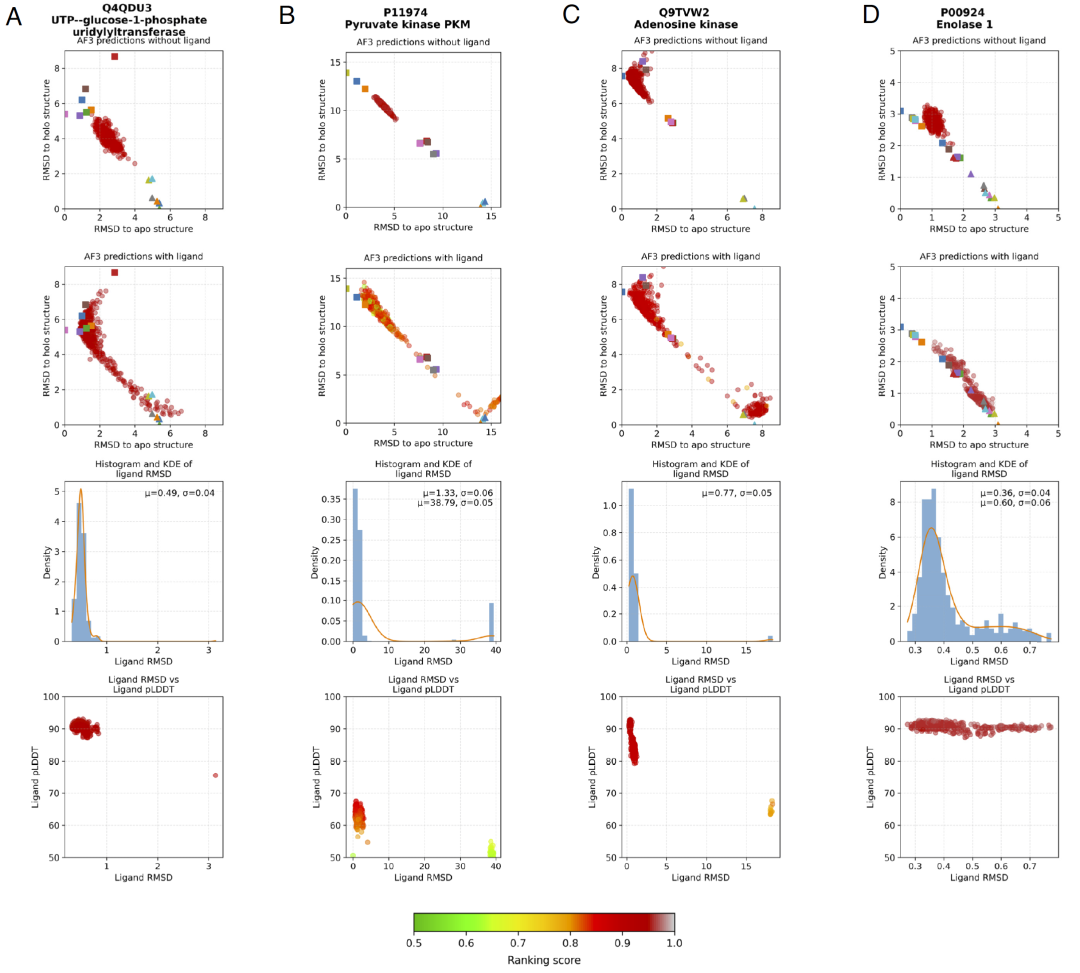
图1: PDB中apo结构多于holo结构的Group 1酶的AF3模型RMSD分布。
AlphaFold3在配体诱导构象变化预测中的偏差
系统比较预测结果后发现,即使输入了正确的配体,模型仍经常预测为训练集中更常见的构象。例如,对于需要明显闭合运动才能形成催化位点的酶,AlphaFold3仍可能预测为开放态。统计分析表明,这种偏差在存在较大结构域旋转或铰链运动时更加明显,说明模型在处理大尺度构象变化方面能力有限。
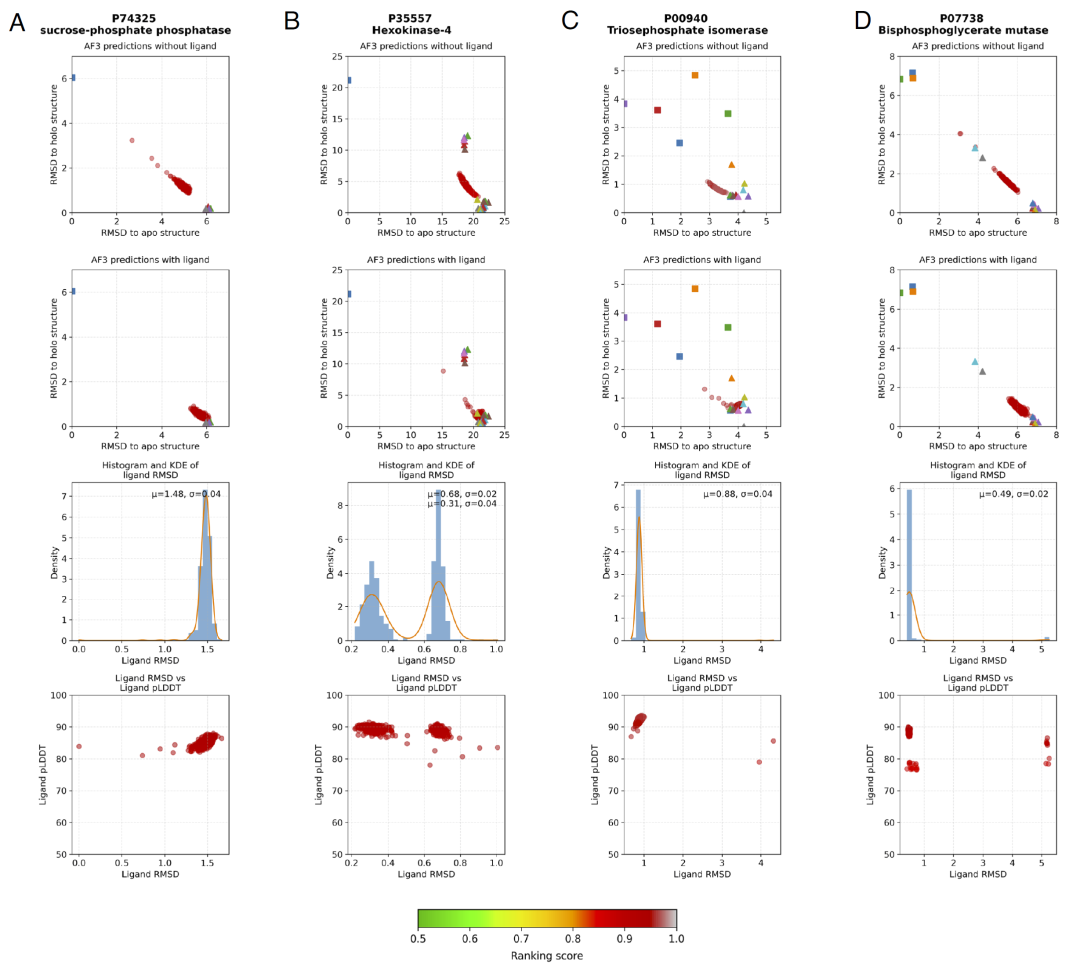
图2:PDB中holo结构多于apo结构的Group 2酶的AF3模型RMSD分布。
模板和训练数据对预测结果的影响
进一步分析发现,当数据库中存在与某一构象高度相似的模板时,AlphaFold3更容易预测为该构象,而不一定符合配体条件。这表明模型在推断结构时仍受到训练数据分布的影响,即使提供了配体信息,也难以完全摆脱对已有结构的依赖。
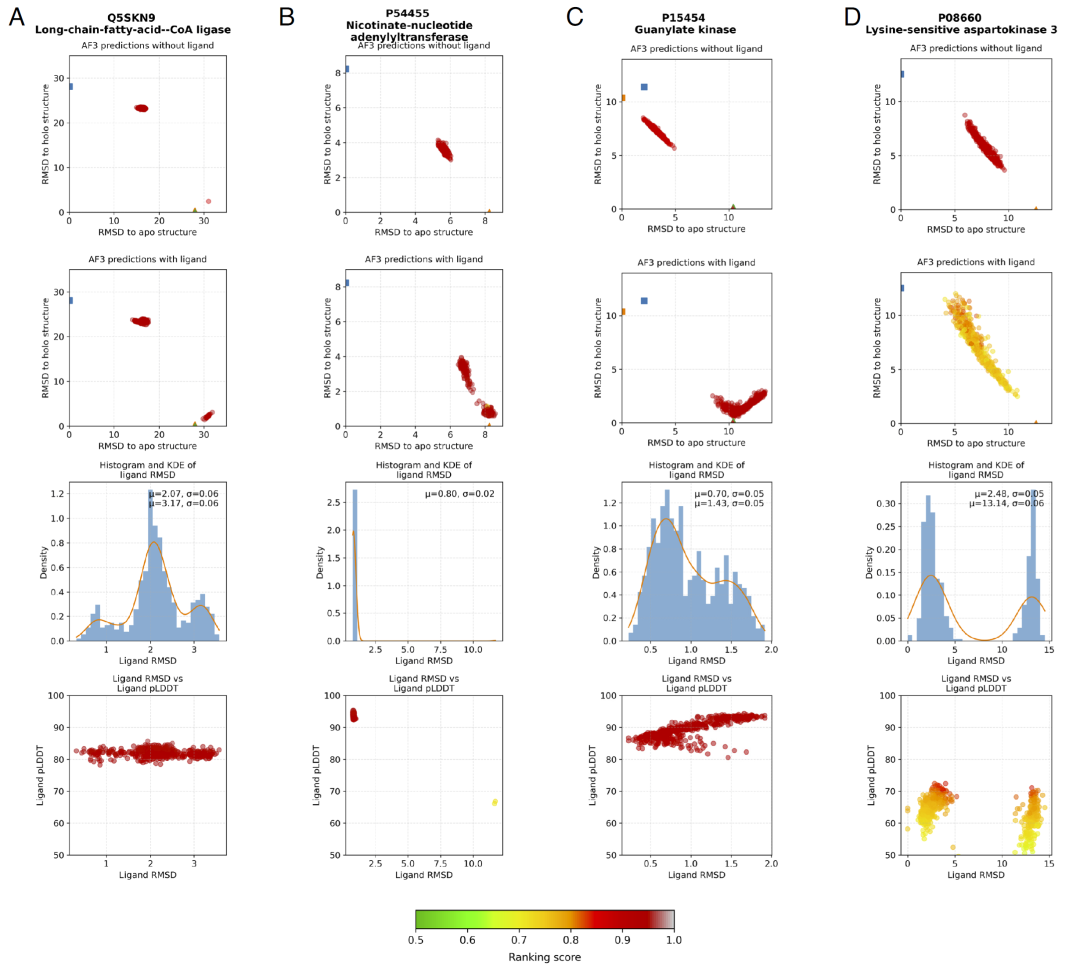
图3:PDB中X射线结构数量极少的Group 2酶的AF3模型RMSD分布。
不同输入策略对预测的影响
研究人员测试了多种输入设置,包括是否提供配体、是否使用模板以及改变序列上下文。结果显示,加入配体可以在部分情况下改善预测,但无法稳定地诱导正确的构象变化。在某些体系中,只有在去除模板或限制模板来源后,模型才更有可能预测到另一种构象,说明模板偏好是重要影响因素。
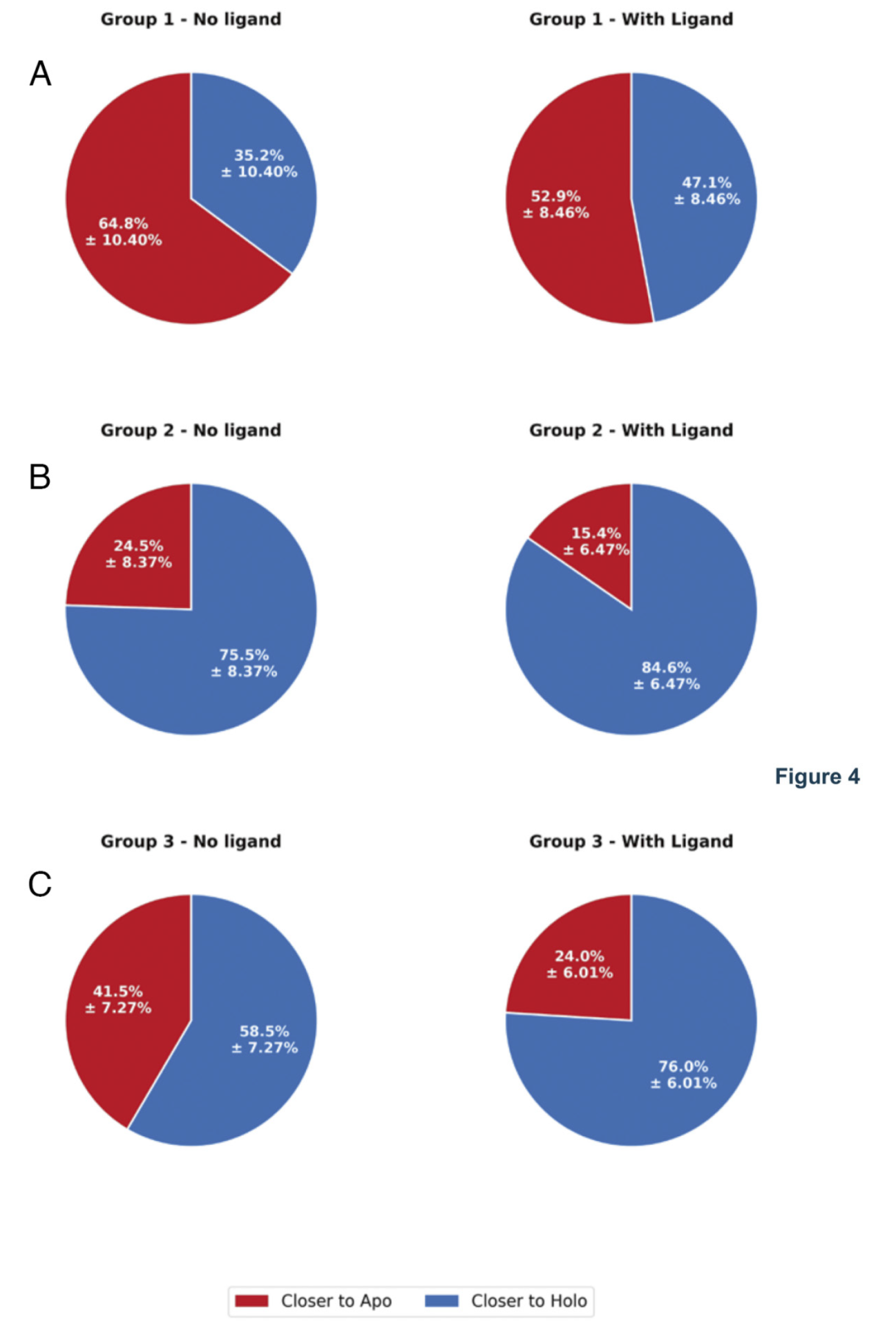
图4:在无配体(左图)和有配体(右图)条件下生成的AF3模型中,更接近apo参考结构(红色)或更接近holo参考结构(蓝色)的模型比例(基于RMSD评估)。
对酶机制研究的影响
在涉及催化闭合或底物诱导构象变化的体系中,如果直接使用AlphaFold3预测结果,可能会得到错误的活性位点结构,从而影响对催化机制的解释。研究人员展示了多个案例,其中预测结构缺少关键的结构域闭合,导致催化残基距离不合理,说明在研究配体调控或动力学机制时需要结合实验或分子动力学方法验证。
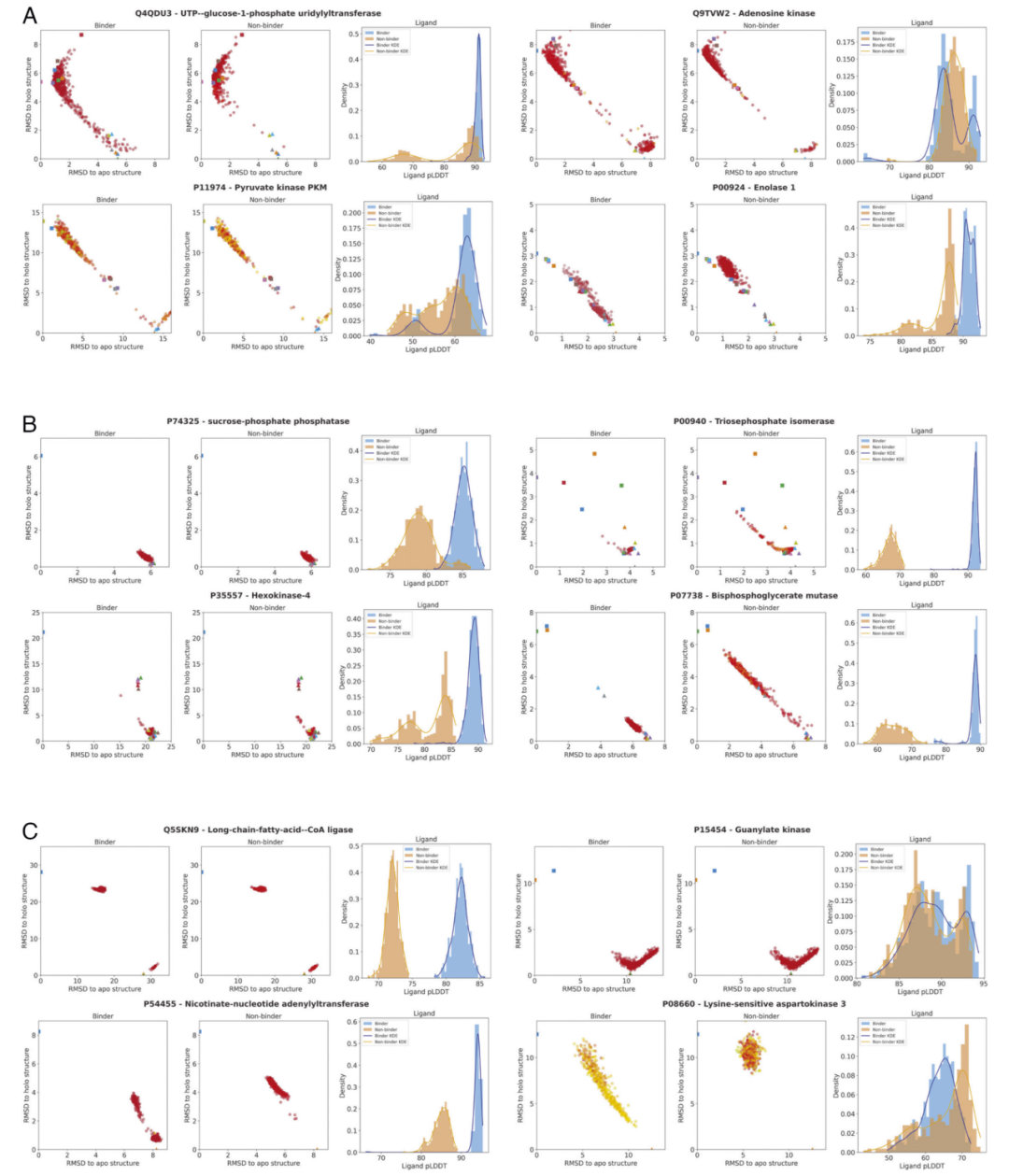
图5:在加入天然配体和非结合配体条件下生成的AF3模型RMSD分布。
讨论
本研究表明,尽管AlphaFold3在静态结构预测方面具有极高精度,但在预测配体诱导的结构域运动时存在系统性偏差。模型倾向于生成训练集中更常见的构象,而不是根据配体条件真实模拟构象转换,这种现象在需要大幅度结构重排的酶中尤为明显。
研究人员认为,这一局限与模型训练数据主要来源于静态晶体结构有关,使得模型更擅长识别稳定构象,而不是模拟能量驱动的构象变化。未来的结构预测方法可能需要结合动力学信息、分子模拟或多构象训练数据,才能更准确地描述蛋白质在功能过程中的运动。
因此,在使用AlphaFold3研究酶催化机制、药物结合或配体诱导构象变化时,应将预测结果视为可能的构象之一,而不是唯一正确结构,并结合实验数据或动力学模拟进行验证。
整理 | DrugOne团队
参考资料
H. Yu,A.A. Bekar-Cesaretli,M. Lazou,D. Kozakov,D. Joseph-McCarthy, & S. Vajda, Bias in the AlphaFold3 prediction of ligand-induced domain motion in enzymes, Proc. Natl. Acad. Sci. U.S.A. 123 (10) e2530709123,
https://doi.org/10.1073/pnas.2530709123 (2026).

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-14,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号