Nat. Commun. | 融合结构建模与深度学习的大肠杆菌蛋白互作组与功能网络构建

Nat. Commun. | 融合结构建模与深度学习的大肠杆菌蛋白互作组与功能网络构建

DrugAI
发布于 2026-04-13 15:50:50
发布于 2026-04-13 15:50:50
DRUGONE
研究人员提出了一种整合多种计算方法的策略,用于在全蛋白组尺度上预测蛋白–蛋白相互作用,并构建功能网络。该方法融合了三类互补信息来源:基于三维结构的PrePPI方法、基于蛋白语言模型的Topsy-Turvy方法,以及基于进化信息评估界面的ZEPPI方法。
在高质量实验数据库的评估中,整合后的模型表现优于任何单一方法,并能够识别更多高置信度相互作用。进一步,研究人员利用AlphaFold3相关算法对部分预测复合物进行结构建模,发现其界面与结构模板模型高度一致。在系统层面,对大肠杆菌互作网络进行聚类分析后,得到数百个功能一致性较高的子网络。这些网络不仅揭示了已知生物过程,还为未知功能蛋白的注释提供了重要线索。

蛋白–蛋白相互作用的预测以及复合物结构建模,是理解细胞功能网络的核心问题。随着AlphaFold等结构预测方法的发展,蛋白结构建模取得了突破,但在全蛋白组尺度上直接预测所有相互作用仍然计算代价极高。
以大肠杆菌为例,潜在相互作用数量可达数百万级,而对于人类蛋白组更是达到数亿级别。即使借助高精度结构预测方法,也难以在可接受时间内完成全局计算。
实验方法虽然在高通量检测方面取得进展,但仍无法覆盖完整互作组,且缺乏结构信息。因此,计算方法成为构建全局互作网络的重要手段。
已有方法通常依赖单一信息源,例如结构同源建模、序列共进化或深度学习预测,但这些方法各有局限。研究人员提出,通过整合不同类型的信息,可以显著提升预测能力与覆盖范围。
方法
研究人员构建了一个基于贝叶斯框架的整合模型,将三种方法的预测结果进行统一融合。
首先,PrePPI通过结构比对,从已知蛋白复合物中寻找模板,将目标蛋白映射到模板结构上,从而构建复合物模型,并通过界面相似性进行评分。
其次,ZEPPI利用进化信息,通过分析界面残基的共进化信号,判断蛋白是否可能形成稳定相互作用。
再次,Topsy-Turvy基于蛋白语言模型,从序列层面预测蛋白是否具有相互作用倾向。
三种方法分别从结构、进化和序列三个维度提供证据。研究人员通过似然比进行整合,使不同来源的信息在统一框架下协同工作,从而得到综合评分。
结果
不同方法的性能比较
研究人员在大肠杆菌高质量互作数据库上评估了各方法的性能。
结果表明,结构方法在低假阳性率区域表现较好,而序列方法具有更高覆盖率。整合方法则在整体性能上优于所有单一方法,同时显著增加了高置信度预测的数量。
特别值得注意的是,不同方法预测的相互作用集合重叠较小,这意味着它们捕获的是互补信息。因此,整合策略能够有效扩展互作网络的覆盖范围。
在严格筛选条件下,整合模型可预测数万条相互作用,其中大部分为此前未被实验记录的新相互作用。
结构预测验证
研究人员选取部分高置信度相互作用,利用AlphaFold3相关方法进行复合物结构预测,并与基于模板的模型进行比较。
结果显示,在多数情况下,两种方法预测的界面高度一致,尤其是在高置信度样本中表现更为显著。这表明结构建模与深度学习方法在界面预测上具有一致性。
此外,一些新预测的相互作用在结构上表现出合理的结合模式,进一步支持其生物学可信性。
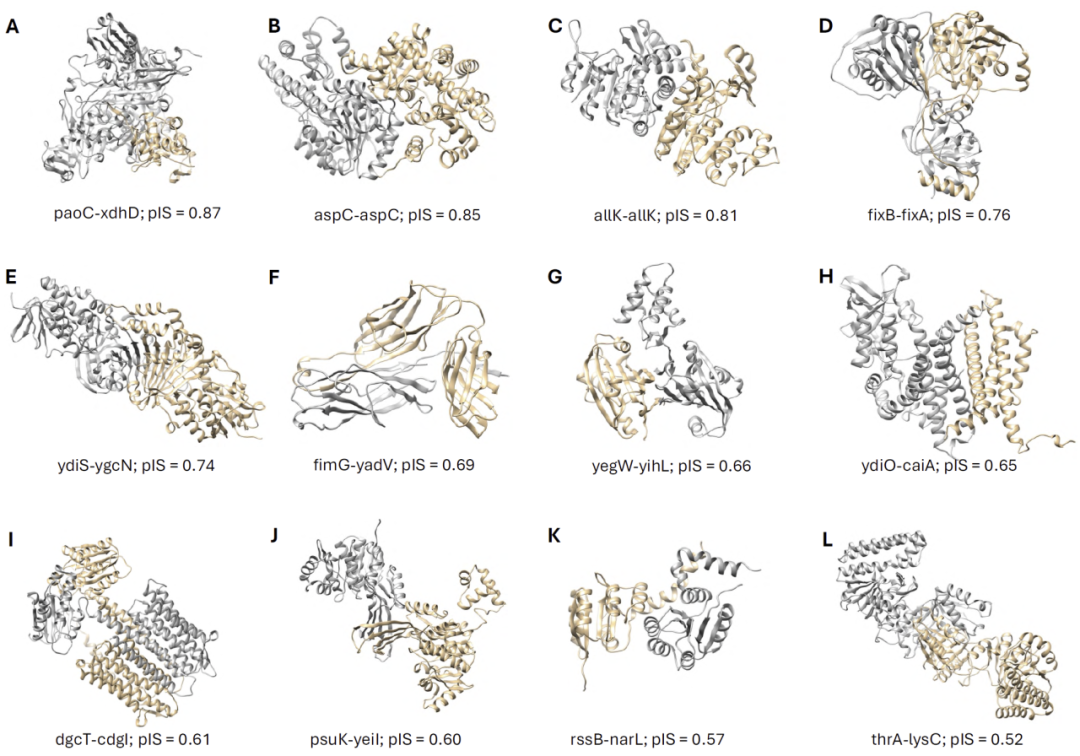
图1:复合物结构预测。
互作网络与功能子网络
研究人员基于预测的相互作用构建了大肠杆菌互作网络,并进行聚类分析。
结果得到数百个子网络,其中大多数具有显著的功能一致性。这一点尤为重要,因为这些网络完全基于预测相互作用构建,并未使用任何功能信息。
通过对这些子网络进行功能富集分析,可以观察到涵盖多种生物过程,例如转录调控、代谢、信号传导以及细胞结构相关过程。
功能网络解析
研究人员进一步分析了若干代表性子网络。在细胞黏附相关网络中,大量蛋白共同参与细胞表面结构形成与稳定,而未注释蛋白也被预测参与这一过程,从而获得潜在功能注释。
在信号转导网络中,多个调控蛋白形成复杂的相互作用关系,揭示了不同通路之间的潜在交叉调控机制。在转录调控网络中,一些未知蛋白被预测与已知转录因子相互作用,提示其可能参与基因表达调控。
在氧化还原稳态网络中,多个蛋白共同维持细胞内的氧化还原平衡,而新预测蛋白可能在该过程中发挥作用。
这些分析表明,互作网络不仅能够重构已知生物过程,还能够为未知蛋白提供功能预测。
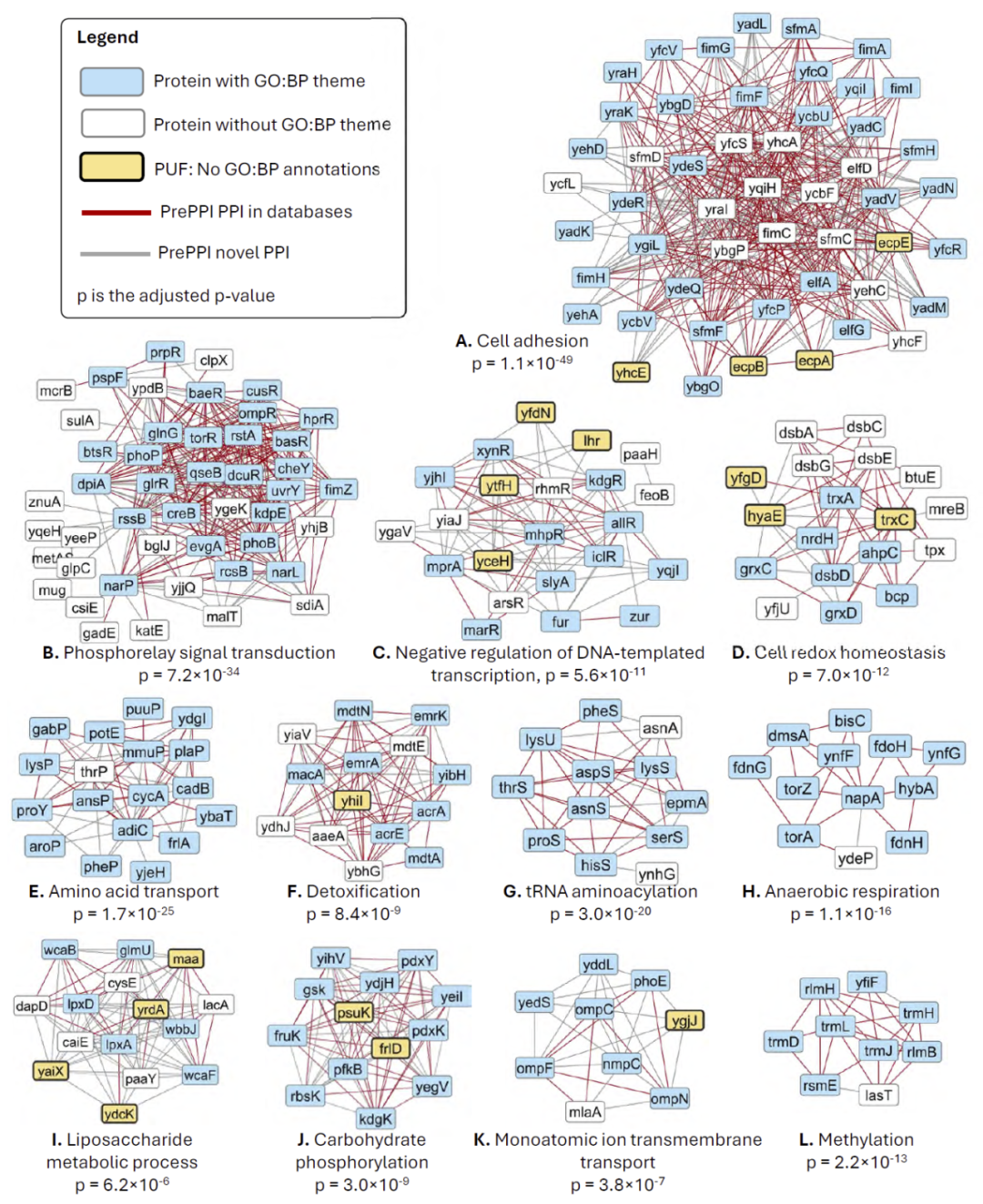
图2:功能子网络。
讨论
研究结果表明,整合多种计算方法能够显著提升蛋白互作预测的准确性与覆盖范围。不同方法之间的低重叠性说明它们捕获了不同层面的信息,而整合策略能够充分利用这些互补优势。
在结构层面,模板建模方法与AlphaFold类方法在界面预测上表现出一致性,这为大规模结构预测提供了可行路径。由于AlphaFold类方法计算成本较高,而PrePPI等方法速度较快,两者可以形成有效的筛选与精细建模流程。
与以往仅关注少量高置信度相互作用的研究不同,本研究更强调构建完整互作网络,从而获得系统层面的生物学理解。这种策略在功能注释和网络分析中具有明显优势。
通过网络聚类分析,研究人员能够在无需先验功能信息的情况下,识别出具有生物学意义的功能模块,并为未知蛋白提供可靠的功能预测。
整理 | DrugOne团队
参考资料
Zhao, H., Velez, C., Naravane, A. et al. Combining structural modeling and deep learning to calculate the E. coli protein interactome and functional networks. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-71166-9

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-12,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号