ATAC-seq实验方案4:升级版Omni-ATAC流程解读(3)-数据分析

ATAC-seq实验方案4:升级版Omni-ATAC流程解读(3)-数据分析

三兔测序学社
发布于 2026-05-13 17:17:15
发布于 2026-05-13 17:17:15
ATAC-seq分析概述
ATAC-seq分析可以分为三个主要步骤:(1)数据处理,(2)峰调用、合并和计数,以及(3)下游分析。
1.数据处理是指从测序仪获取原始FASTQ文件,并准备将它们与基因组比对,同时调整Tn5偏移量
2.处理后的读段将用于逐一样本进行call peaks 招募峰。然后,讲所有样本的峰进行合并,形成联合峰集,并计算每个联合峰集峰数量。
3.下游分析,包括差异可及性分析(确定染色体DNA上哪些峰显著更加或更少可及开放),以及在相关的峰集上的转录因子印迹(motif)模体富集。
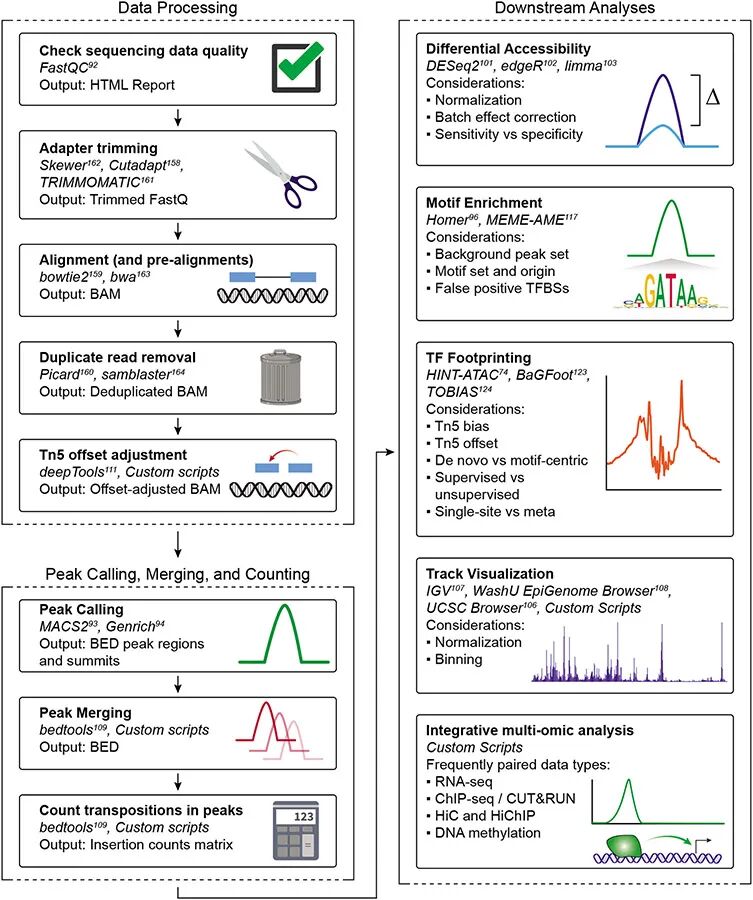
1.ATAC-seq的最测序结果通常是FASTQ格式文件,每个文库包含双端测序的Read 1和Read 2文件,对应ATAC-seq片段两端的配对末端读段。每个读段都是由一个n5转座事件产生的。可以使用FastQC来检查每个文库的整体测序质量,它提供一个HTML报告,包含每个碱基的质量评分以及数据中任何观察到的偏差。
2. 标准的ATAC-seq流程会以FASTQ文件为输入,执行一系列质量控制和数据清理步骤,然后与参考基因组进行比对。首先,会移除任何与ATAC-seq接头序列对应的碱基,ATAC-seq片段可能比测序读长短,导致读段末端读入接头序列。由于这个外源序列会干扰正确的对齐,必须移除。来自线粒体DNA的读段(由于转座到非染色质化的线粒体基因组而产生,被认为是“污染”的)应该在全基因组比对之前或之后移除。一般来说,使用Omni-ATAC方案,我们预计看到少于15%的读段映射到线粒体基因组。更高的百分比可能表明过度裂解或在使用ATAC-seq洗涤缓冲液时未充分移除线粒体,可能需要以更高深度测序文库,以获得足够的唯一读段映射到核基因组。剩余的读段映射到参考基因组,并过滤掉低质量的对齐。
3. 接下来,根据序列片段的相同起始和结束位置标记并移除PCR重复。一般来说,我们发现使用这个方案从50,000个细胞生成的ATAC-seq文库需要最小的扩增(3-7个额外循环)。这个扩增水平与文库复杂性相关联,通常导致少于10%的对齐读段被标记为重复;然而,重要的是要注意,标记为重复的读段百分比取决于许多因素,包括总测序深度,我们不使用这个指标来确定文库质量。例如,含有大量死细胞的样本会导致文库复杂性高且重复读段百分比低,因为它们的DNA大部分去染色质化,因此完全可及于转座酶;然而,这不被认为是高质量的ATAC-seq文库。尽管如此,高重复读段百分比(>25%),特别是当文库由50,000个细胞制成并测序到约10百万读段对时,可能表明转座效率低下,可能反映输入材料不足或质量差。我们发现大多数ATAC-seq文库在这一系列质量控制步骤后,有>80%的总读段映射到细胞核基因组。如上所述,TSS富集分数是最重要的质量控制指标,应在深度测序数据上重新计算。
4. 最后,调整ATAC-seq片段的“Tn5偏移”。这个Tn5偏移考虑了Tn5转座酶二聚体的分子机制。Tn5以同源二聚体形式结合到DNA,两个Tn5分子之间有9个碱基的DNA段。因此,每个同源二聚体结合事件产生两个插入,相隔9个碱基,中间的9个碱基被复制并存在于相应的片段上。这个Tn5结合事件的真正中心点在这个9碱基区域的中间。考虑这一点,已经采用调整每个读段起始位置的惯例,通过向正链插入添加4个碱基,向负链插入添加−5个碱基。
峰调用、合并、计数和注释:
比对后,下一步分析是识别染色质可及性的“峰”——即在基因组中富集Tn5插入的区域,这表明样本中的许多细胞在此位置具有Tn5可及的染色质。这些峰的位置,以及在某种程度上它们的大小,可以帮助我们理解基因调控的整体情况。
有许多软件可以识别这些峰区域,尽管MACS2和最近的Genrich是最常用的两个。HMMRATAC专门设计用于ATAC-seq数据的峰调用,Genrich也提供了ATAC-seq专用模式。由于MACS2是ATAC-seq流程中最常用的峰调用工具,我们在这里重点介绍它。MACS(Model-based Analysis of ChIP-Seq)是一种流行的峰调用工具,适用于各种数据集,包括ChIP-seq、ChIC-seq、DNAse-seq和ATAC-seq。MACS2使用滑动窗口方法调用峰,用户提供的窗口大小沿基因组滑动,以找到相对于背景信号具有倍数富集(也由用户提供)的区域。因为MACS2并不是专门为ATAC-seq设计的,某些参数调整是至关重要的。例如,为了在MACS2中准确表示单个Tn5插入事件,应使用“shift”和“extsize”参数,以确保表示Tn5插入的单碱基位置直接位于提供给MACS2的“读段”的中间。为此,上述PEPATAC流程在MACS2中进行峰调用,默认设置如下:--shift −75 --extsize 150 --nomodel --call-summits --nolambda --keep-dup all -p 0.01。需要注意的是,MACS2中的峰调用受测序深度的影响很大。因此,数据集中的读段越多,调用的峰也越多,每个峰的显著性也越大。因此,不适合在样本之间比较原始峰调用。过去,我们建议使用标准化峰得分,可以通过将单个峰得分(−log10(p-value))标准化到样本中识别的所有峰得分的总和来获得。
峰集应首先在特定样本和可能的技术重复基础上进行识别,然后在所有将要比较的样本之间进行合并,创建我们所说的联合峰集。例如,如果将处理化合物的细胞与处理载体对照的细胞进行比较,应首先为每个技术重复调用峰,然后在组内的所有技术重复之间合并(例如,所有载体对照),最后在两个实验组之间合并(例如,处理和对照)。虽然这种峰合并听起来很简单,但有许多调用和合并峰的方法,业内尚未达成共识。我们建议使用以峰顶为中心的不重叠固定宽度501-bp峰作为标准,因为大多数DNA调控元件的长度小于500-bp。固定宽度的峰使下游计算更容易,因为不需要标准化峰长度,而变量宽度峰的方法则需要。为了在样本之间合并峰,我们建议使用一种迭代重叠过程,该过程保持固定峰宽度,同时避免偏向深度测序的文库或不同样本类型的重复数量差异。在迭代重叠中,首先根据其标准化显著性(基于MACS2评分)对峰进行排名,然后保留最显著的峰,并迭代地移除任何重叠的峰,直到没有重叠的峰为止。迭代重叠避免了峰合并中的两个常见问题:1)在比较多个样本时,由于每个样本特定的峰集不会完全重叠,创建越来越大的峰;2)当使用聚类重叠技术时,由于将所有聚集在一起的峰保留一个赢家来总结聚类,导致灵敏度下降。为了便于使用这种迭代重叠峰合并方法与任何一组MACS2峰调用,我们提供了在以前研究中使用的峰合并脚本(https://github.com/corceslab/ATAC_IterativeOverlapPeakMerging)。
一旦创建了联合峰集,每个峰每个样本的转座事件数量将被编译成一个插入计数矩阵。这个原始矩阵应根据峰内的总读数进行标准化,然后进行对数变换,尽管这些操作通常作为标准差异分析工作流的一部分执行。通过将峰注释到最近的基因,可以部分推断峰的功能。有多种工具可以执行此分析,包括HOMER(HOMER annotatePeaks.pl)、ChIPseeker和ChIPpeakAnno。虽然最近的基因提供了一种简单的方法来注释峰的功能,但重要的是要记住,远端调控元件可以在大的基因组距离上起作用,最近的基因通常不是每个峰的真正目标。
评估样本和技术重复的一致性:
获得插入计数矩阵后,应评估技术或生物重复之间的相关性。生物重复的精确Pearson相关系数在很大程度上取决于重复的来源。例如,可以期望从三只同一近交背景的小鼠中分离的CD4+ T细胞的Pearson相关值大于0.9,而从三个不同的人类供体中分离的相同细胞类型的Pearson相关值可能大于0.8,这是由于人类之间的自然变异比共同饲养的近交小鼠大。无论样本来源如何,我们期望技术重复(即在同一时间使用不同批次的相同起始材料进行的两个ATAC-seq反应)的Pearson相关性大于0.9,理想情况下大于0.95。这些样本之间的关系通常使用一对一图或Pearson相关图进行可视化。
差异可及区域:
可以通过多种方式识别差异可及区域(DARs)。选择哪种工作流和工具用于差异可及性测试应基于项目的目标,以及将进行多少验证或后续分析。一般来说,Gontarz等人的结果建议DESeq和DESeq2是希望最小化假阳性(即最小化误发现率)的研究人员的最佳选择。相反,对于目标是尽可能检测到更多潜在DARs并容忍假阳性的应用,edgeR或limma是高灵敏度的良好选择,其中limma在信号较低的区域(如远端增强子)中给出了最佳结果。高灵敏度技术可能对利用与其他测序技术的综合分析的研究有用,这可以为DARs提供额外的信心。
批次效应:
批次效应可能在ATAC-seq多个步骤中发生,并可能影响下游分析,特别是在比较不同时间点或不同组获得的ATAC-seq数据集时。通常,在特定实验背景下生成ATAC-seq数据时,应使用最佳实践规则,以限制由技术伪影引起的变异。然而,一些批次效应的来源可能难以控制,因为它们也可能源自上游样本属性,例如人类组织的死后间隔时间。为了消除不同ATAC实验之间这些不必要的变异来源,可以使用edgeR或DESeq2中的内置批次校正工具。这两个程序都允许在实验设计中包含批次作为协变量。在edgeR中,批次通过负二项式广义线性模型(GLM)回归出,然后执行似然性检验作为配对样本t检验的广义化。类似地,DESeq2也适配负二项式GLM,并使用Wald检验来确定实验条件的显著性。除了这两个程序,还有RUVseq,主要用于RNA-seq数据,但也已应用于ATAC-seq数据。它对由edgeR计算的残差进行因子分析,使用上四分位数标准化计数。
可及性峰可视化:
无论使用哪种工作流识别DARs,我们建议使用正确标准化的测序轨迹对重要峰进行视觉检查。这确保了通过差异分析识别的差异符合预期。一种常见的方法是创建标准化的bigWig(.bw)文件,可以上传到基因组浏览器工具,如UCSC基因组浏览器、集成基因组浏览器(IGV)或华盛顿大学表观基因组浏览器。bigWig文件提供了一种简化的方式来可视化基因组范围的对齐,在ATAC-seq数据的情况下,基因组范围的染色质可及性模式。可以使用bedtools genomecov或deepTools bamCoverage进行标准化,应用基于TSS区域读数的比例因子。这种方法类似于基于峰读数的标准化,使得可以同时对测序深度和数据质量进行标准化。我们建议使用TSS区域的读数,因为这些区域在样本之间是不变的,而使用峰读数需要在峰集更改时重新标准化。我们还建议在创建bigWig文件时使用标准化的bin大小。bin数值越大,分辨率越低,生成的bigWig文件越小。我们建议大多数应用使用100 bp的bin大小。
模体Motif富集分析:
模体富集分析基于在给定峰集中搜索转录因子结合模体。模体信息,包括实验观察和计算预测,已汇总在多个大型数据库中,如JASPAR、CIS-BP和ENCODE。每个模体存储为位置权重矩阵(PWM),多种工具可以扫描每个峰的DNA序列以查找每个模体的存在,包括HOMER、TFBSTools、motifmatchr和MEME。在给定峰集中相对于背景区域集的模体存在的富集,意味着该TF可能在驱动该峰区域的可及性方面起着重要作用。例如,在比较分化细胞和未分化细胞时,这些模体富集工具将识别在分化后获得的峰中统计显著富集的模体,与分化细胞和未分化细胞共享的模体相比。这可以提供关于驱动分化的TF的假设。已经使用多种统计检验来识别模体富集,包括超几何检验(HOMER)和秩和检验(MEME)。在进行这些分析时,重要的是选择一个有意义的区域或峰集作为富集检验的背景。通常不正确使用整个基因组作为背景,应使用匹配的峰子集或整个峰集,具体取决于应用。
转录因子足迹分析:
转录因子(TF)对可及峰的调控也可以通过足迹分析来确定。足迹分析起源于经典的DNase足迹法,后来被适应于基于测序的分析(有时称为数字基因组足迹法)。在ATAC-seq的背景下,TF足迹会在TF结合到DNA并防止Tn5插入特定直接结合的DNA碱基时出现。这留下了一段DNA,或“足迹”,其中插入比相邻的无核小体(和无TF)区域少得多(图4)。因此,足迹算法的目标是首先识别这些“凹陷”Tn5插入的位置,其次,通过检查推断直接结合的中心碱基序列,确定哪个TF可能结合在那里。识别TF足迹可以帮助重建基因调控网络,并可能比模体扫描更具体地检测TF的存在。然而,将足迹分析应用于ATAC-seq数据存在许多挑战,解释往往会因Tn5的序列偏差而复杂化。经典上,TF足迹设计用于预测基因组中单个位点的TF结合。这需要许多片段映射到特定位点,以提供足够的观察来识别受保护的碱基。在对50,000个细胞进行ATAC-seq时,通常无法获得足够的深度来进行单位点足迹分析,即使在高测序深度下也是如此。因此,许多研究人员进行了“元”足迹分析,其中数百或数千个包含特定TF模体的基因组位点被汇集成一个足迹。需要注意的是,这不是经典的足迹使用方法,其结果与模体富集更相似,而不是单位点足迹。与其他ATAC-seq方法一样,TF足迹分析应调整片段以适应Tn5偏移,并考虑Tn5插入偏差。有关单位点足迹分析的几种方法,包括HINT-ATAC、BaGFoot和TOBIAS,在补充说明4中有讨论。
核小体定位:
ATAC-seq数据也被用于研究核小体定位,尽管这些技术仍在开发中,需要非常高的测序深度才能准确计算。核小体定位是指尝试测量某些位点核小体组织的差异,包括它们的位置和占有率。核小体分布的变化与给定启动子的基因调控和表达水平的变化有关。经典上,核小体定位是使用MNase-seq数据进行的,并为此类数据开发了几种工具。然而,NucleoATAC和HMMRATAC专门用于从ATAC-seq数据预测核小体位置。一般来说,对于平均ATAC-seq文库的测序深度,不建议应用核小体分布技术。
将ATAC-seq与其他‘组学’分析技术结合:
如介绍中所述,ATAC-seq提供了基因组范围的染色质可及性的整体景观。因此,将ATAC-seq的染色质可及性景观与其他匹配的基因组、转录组或表观基因组分析结果进行比较可能是有趣的。多组学可能有助于解释给定ATAC-seq峰的功能。例如,可以将ATAC-seq与H3K27ac ChIP-seq(或其他ChIC技术)联合分析,预期ATAC-seq峰的一部分将与H3K27ac标记的活跃增强子和启动子重叠。有足够大的数据集,跨样本的ATAC-seq染色质可及性与附近基因表达之间的相关性可以用于识别代表潜在基因调控相互作用的峰与基因的联系。这些相互作用可以通过染色质构象捕获技术(如HiC或HiChIP)进行正向验证。
类似地,ATAC-seq提供了一个独特的窗口,以研究非编码基因组中序列变化的影响。通过结合ATAC-seq数据与全基因组测序数据,可以识别等位基因不平衡区域,其中两种不同等位基因显示出不同的可及性,暗示不同的TF结合。这已被用于识别染色质可及性量率位点,并开始为通过基因组范围关联研究识别的非编码多态性赋予功能。这些类型的分析可以帮助为通过ATAC-seq识别的特定潜在调控元件赋予相关性或功能。
准备发表:发表中包含的ATAC-seq数据应存储在公共可访问的存储库中,如基因表达全集(GEO)、测序读取存档(SRA)或欧洲核苷酸存档(ENA)。每个存储库的具体说明可以在其网站上找到。然而,最常见的是包括以下文件:1)所有原始FASTQ文件(Read 1和Read 2)在QC和接头修剪之前,2)所有分析样本的联合峰集的插入计数矩阵,包括峰的基因组坐标,3)可以在基因组浏览器上轻松可视化的标准化bigWig文件,以及4)如果适用,差异可及区域(DARs)的列表。我们还鼓励研究人员包含一个补充表,报告每个测序样本的QC指标,特别是TSS富集分数。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-10,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号