JMC | 候选药物诞生之前,分子经历了什么?

JMC | 候选药物诞生之前,分子经历了什么?

MindDance
发布于 2026-05-20 13:37:43
发布于 2026-05-20 13:37:43
一项药物发现项目从命中化合物走到临床候选药物,分子到底发生了什么变化?发表于 Journal of Medicinal Chemistry 的这篇长文,做了一次相当扎实的回顾:作者 Paul D. Leeson 手工梳理了 2018—2024 年药物化学文献中成功进入候选药物阶段的优化案例,主分析集中在 487 组起始化合物—候选药物配对,其中 469 组同时有体外活性数据。
Leeson 不是纯粹的数据分析作者。他有四十多年药物发现经验,曾在 MSD、阿斯利康和 GSK 工作,长期关注化合物质量、药物样性和药物化学决策。这篇文章的底色也很明显:它不是把优化过程简化成几个机器学习特征,而是在问一个更贴近药化现场的问题——成功优化的分子,通常是怎样被长出来、修出来、压住风险的。
核心答案:成功优化通常会让分子变大、变复杂、活性提高,但并不会让平均脂溶性继续上升。候选药物的平均分子量比起始化合物高出约 81 Da,体外活性平均提高 1.5 个对数单位,约等于三十倍量级;与此同时,XLogP3 从 3.1 到 3.0,几乎没有增加。换句话说,现代先导优化已经不是简单地往分子上加疏水基团,而是在分子增长中同时守住脂溶性、氢键供体、芳香性和可开发性。
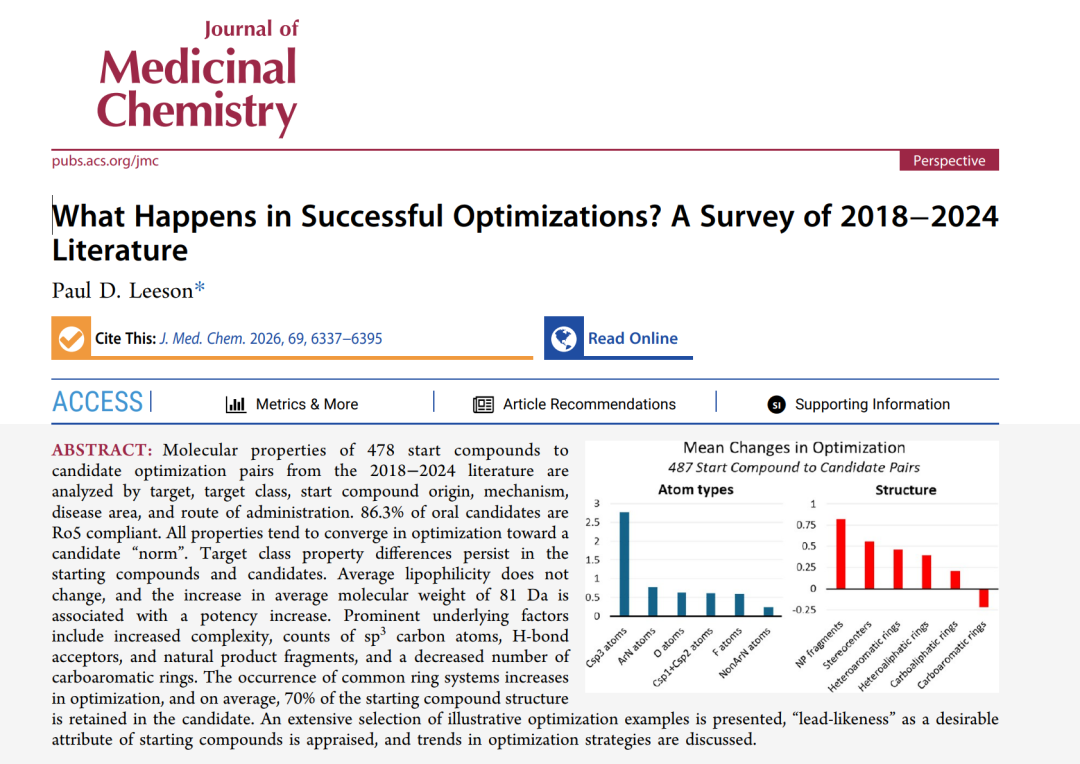
本研究的数据底座:文献来源、专利时间、起始化合物来源、靶点类别、疾病领域、作用机制和给药途径分布。
本研究的数据底座:文献来源、专利时间、起始化合物来源、靶点类别、疾病领域、作用机制和给药途径分布。
先导优化的老问题,到了今天并没有变简单
药物化学里有一个很朴素、也很难的问题:一个早期命中化合物,怎样才能变成真正有希望进入临床的候选药物?
早期命中化合物往往只是说明某个分子能够影响某个靶点。它可能活性不够,代谢太快,溶解度差,暴露不稳定,选择性不够,也可能在细胞或动物体内完全表现不出体外实验里的漂亮数据。先导优化要做的事情,就是在这些彼此牵制的指标之间找平衡。药物化学家常说的多参数优化,真实含义并不是把每个参数都做到漂亮,而是在项目允许的边界内,让分子够好、够稳、够可开发。
这个问题从 1990 年代后期就被系统讨论过。当时提出的先导样性思想认为,理想的筛选起点不应太大、太脂溶。经典标准大致是分子量小于 350、logP 小于 3,活性不必一开始就很强。背后的逻辑很清楚:小分子还有生长空间,后续可以通过增加片段、改善结合、提高活性,同时把代谢和药代问题慢慢修正。片段药物发现也是类似思路,只是把起点进一步缩小到分子量约 175—230 的片段区间。
但过去二十多年,靶点空间发生了很大变化。越来越多项目面对的是蛋白—蛋白相互作用、变构位点、蛋白降解、共价调控、宏环或超越五规则空间。这些靶点常常要求更大的结合表面积、更复杂的构象控制,也就意味着分子天然更大、更极性、更难口服。传统先导样性还能不能直接照搬,是一个需要重新审视的问题。
这篇文章放在这个语境里,价值就不只是更新一组统计数字。它把 2018—2024 年公开文献里近五百个成功优化案例放到一起,看不同来源的起点、不同靶点类别、不同疾病领域、不同给药途径,最终是否会走向相似的候选药物空间。结果显示:起点可以不同,路线可以不同,但成功候选药物确实会向某种候选药物常态收敛。
数据从哪里来:不是数据库一键下载,而是逐篇文献筛出来
作者收集的是 2018—2024 年药物化学文献中被明确描述为进入临床、准备进入临床,或已完成较深入临床前评估且优化路径清楚的候选药物案例。数据并不是简单关键词检索得到,而是逐篇阅读文献后人工整理。
论文共识别到 498 个候选药物,其中 487 个可以找到明确的化学起点,形成起始化合物—候选药物配对;469 组同时有起点和候选物的体外活性数据。候选药物发表时间通常滞后于真实发明时间,文章统计显示,候选药物相关专利优先权多集中在 2011—2016 年前后,平均约在 2014 年。也就是说,这篇 2026 年文章看到的成功,很多其实是十年前项目决策的结果。
起始化合物来源大致分为六类:已有药物、已知文献化合物、药效团假设设计、聚焦筛选、片段筛选和多样性筛选。其中,已有药物占 12%,已知化合物占 27%,药效团假设占 7%;聚焦筛选占 12%,片段筛选占 5%,多样性筛选占 37%。如果粗略合并,来自已知知识体系的起点约占 46%,来自筛选策略的起点约占 54%。这说明成功候选药物并不只来自高通量筛选,也不只来自老分子改造,二者在当前公开案例中几乎各占半壁江山。
靶点方面,激酶占 28%,其他酶类占 22%,GPCR 占 15%;疾病领域中,肿瘤占 34%,中枢神经系统占 17%,感染占 11%。给药途径上,口服候选药物占绝对多数,达到 89%。这些比例决定了这篇文章的结论主要代表传统小分子药物化学空间,而不是完全代表 PROTAC、分子胶、宏环肽等新模态空间。
这里有一个很重要的边界:这不是失败项目全集,也不是企业内部完整项目数据库。它看到的是已经被公开发表、并且有清晰优化路径的成功案例。因此,它回答的不是所有优化为什么失败,而是已经成功的优化,留下了哪些可重复观察的分子变化痕迹。
第一层结论:分子会长大,但脂溶性被看住了
文章最直接的统计结果可以概括为一句话:成功优化往往伴随分子增长,但不是脂溶性失控式增长。
在 487 组配对中,候选药物相对起始化合物的平均分子量增加 81 Da,重原子数增加 5.7,氢键受体增加 2.0,拓扑极性表面积增加 17.6 Ų,可旋转键增加 1.1,氟原子数增加 0.6。体外活性平均提高 1.5 个对数单位。与此同时,脂溶性指标基本没有上升:XLogP3 平均从 3.1 到 3.0,clogP 从 3.2 到 3.0,logD 维持在 2.7 左右。氢键供体也几乎不变,平均只从 1.78 到 1.90。
关键性质 | 起始化合物均值 | 候选药物均值 | 平均变化 | 解读 |
|---|---|---|---|---|
分子量 MW | 404 | 485 | +81 Da | 分子增长是常态 |
XLogP3 | 3.1 | 3.0 | -0.1 | 平均脂溶性没有增加 |
体外活性 p(activity) | 6.9 | 8.4 | +1.5 | 活性约提升三十倍量级 |
氢键受体 HBA | 5.06 | 7.08 | +2.02 | 极性主要靠受体增加 |
氢键供体 HBD | 1.78 | 1.90 | +0.11 | 供体被严格控制 |
脂溶性配体效率 LLE | 3.7 | 5.3 | +1.6 | 活性提升不是靠堆脂溶性 |
sp³ 碳原子数 | 6.78 | 9.55 | +2.77 | 三维性和脂肪性增强 |
碳芳香环数 | 1.48 | 1.26 | -0.22 | 苯环类负担被削减 |
杂芳香环数 | 1.26 | 1.72 | +0.46 | 含氮杂芳环更常见 |
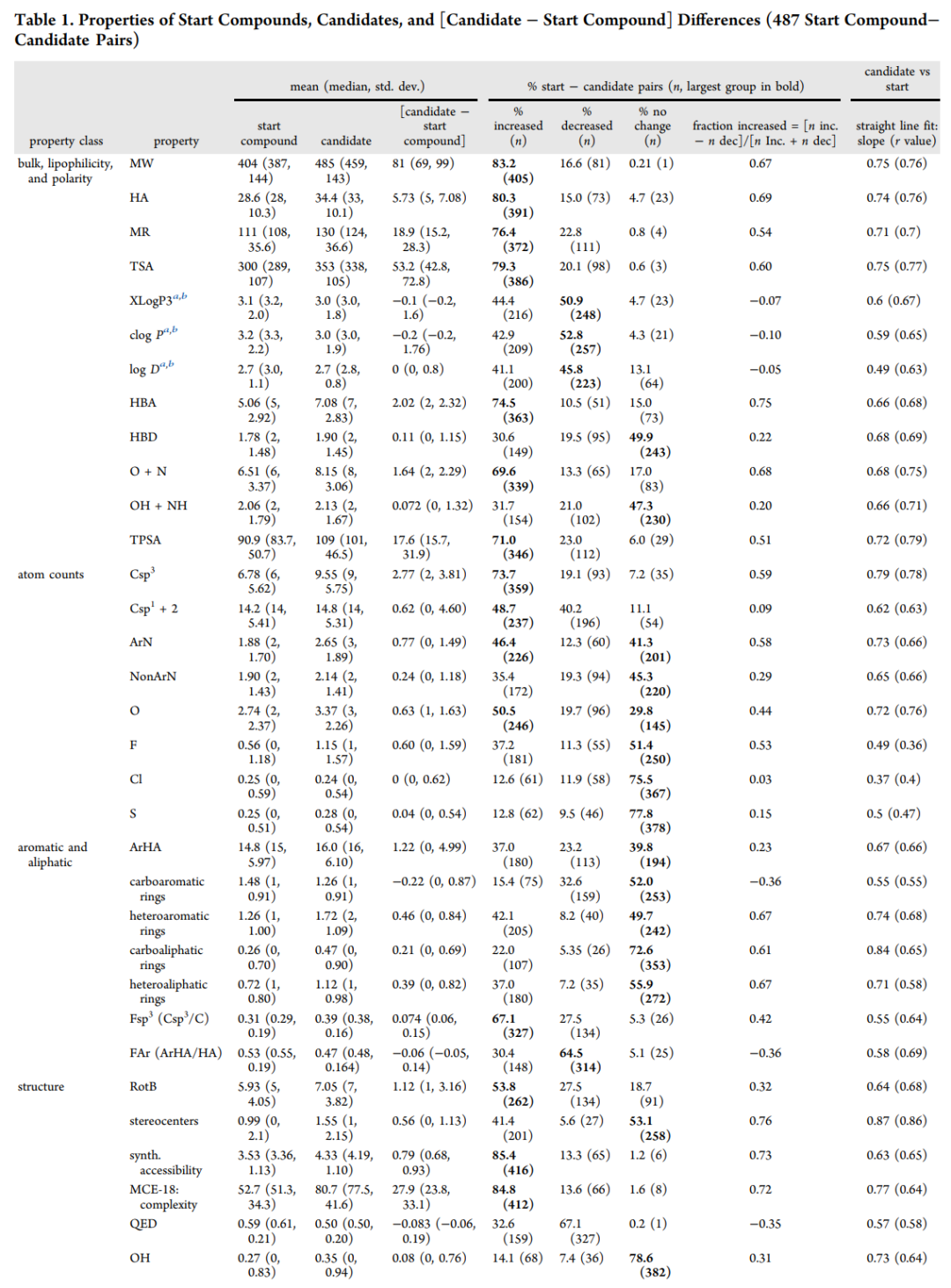
起始化合物与候选药物的关键物化性质变化。分子量、氢键受体、sp³ 碳和活性显著增加,而脂溶性和氢键供体基本保持稳定。
起始化合物与候选药物的关键物化性质变化。分子量、氢键受体、sp³ 碳和活性显著增加,而脂溶性和氢键供体基本保持稳定。
这种变化在药化实践中很有画面感。为了让分子更好地占据结合口袋,药化团队往往会增加片段、延伸取代基、引入新的环系或立体中心。分子变大以后,范德华接触增加,活性自然更容易提升。但一旦靠增加脂溶性来换活性,代谢清除、溶解度、非特异性结合和安全性问题就会很快追上来。因此,真正的优化不是单纯把分子做大,而是在分子长大的同时,用极性、杂原子、环系和构象约束把脂溶性压住。
这也是 LLE 增加而 LE 基本不变的原因。LE 按重原子数归一化,分子变大以后活性提高会被重原子增加抵消;LLE 则直接反映活性与脂溶性的差值。文章中 LLE 平均从 3.7 提高到 5.3,说明成功候选药物的活性提升并不是靠无节制增加脂溶性买来的。
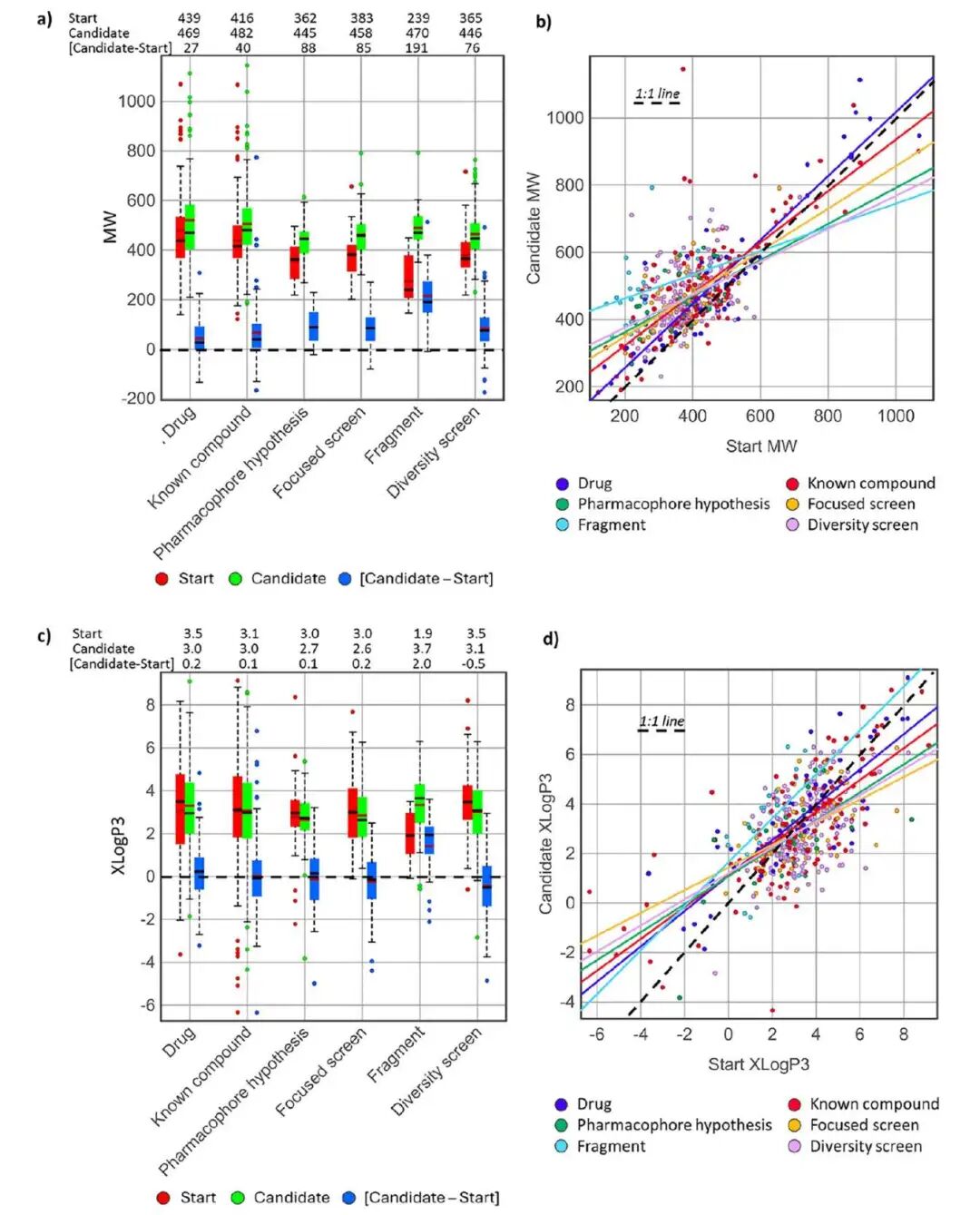
不同起点来源的优化路径:候选药物普遍走向相近分子量区间,活性提升与分子增长同步发生,但脂溶性整体受到控制。
不同起点来源的优化路径:候选药物普遍走向相近分子量区间,活性提升与分子增长同步发生,但脂溶性整体受到控制。
第二层结论:真正被优化出来的是复杂度、杂原子和更可控的环系
如果只看分子量增加,容易把结论读浅。文章进一步把重原子增加拆开,发现平均新增的 5.73 个重原子中,98% 可以由六类原子变化解释:sp³ 碳增加 2.77,芳香氮增加 0.77,氧增加 0.63,sp²/sp¹ 碳增加 0.62,氟增加 0.60,非芳香氮增加 0.24。
这里面最重要的不是碳原子总数,而是sp³ 碳和芳香氮。sp³ 碳增加,往往意味着更多脂肪环、更多立体中心、更强构象控制,也就是药化中常说的三维性增强。芳香氮增加,则常常意味着用吡啶、嘧啶、吡唑等杂芳环替代苯环,在保留结合能力的同时降低脂溶性,改善溶解度或代谢风险。
文章还观察到一个很清楚的芳香环趋势:碳芳香环平均减少 0.22,杂芳香环平均增加 0.46。换句话说,现代候选药物并不是远离芳香性,而是从苯环主导的芳香性转向杂芳环参与的芳香性。这很符合药化现场的经验:苯环是可靠的结合元件,但苯环多了以后,平面性、结晶堆积、脂溶性和代谢风险都会变得难处理。杂芳环和脂肪环替代,正是许多项目后期反复打磨的方向。
氟原子的变化也很显眼。含氟化合物在起始化合物中占 25.9%,在候选药物中升至 50.3%。这不是简单的流行元素偏好。氟可以微调脂溶性、改变代谢软点,有时还能改善活性或选择性。相比氯,氟对脂溶性的推高通常更温和,因此在后期精修中更容易成为药化团队手里的细刀。
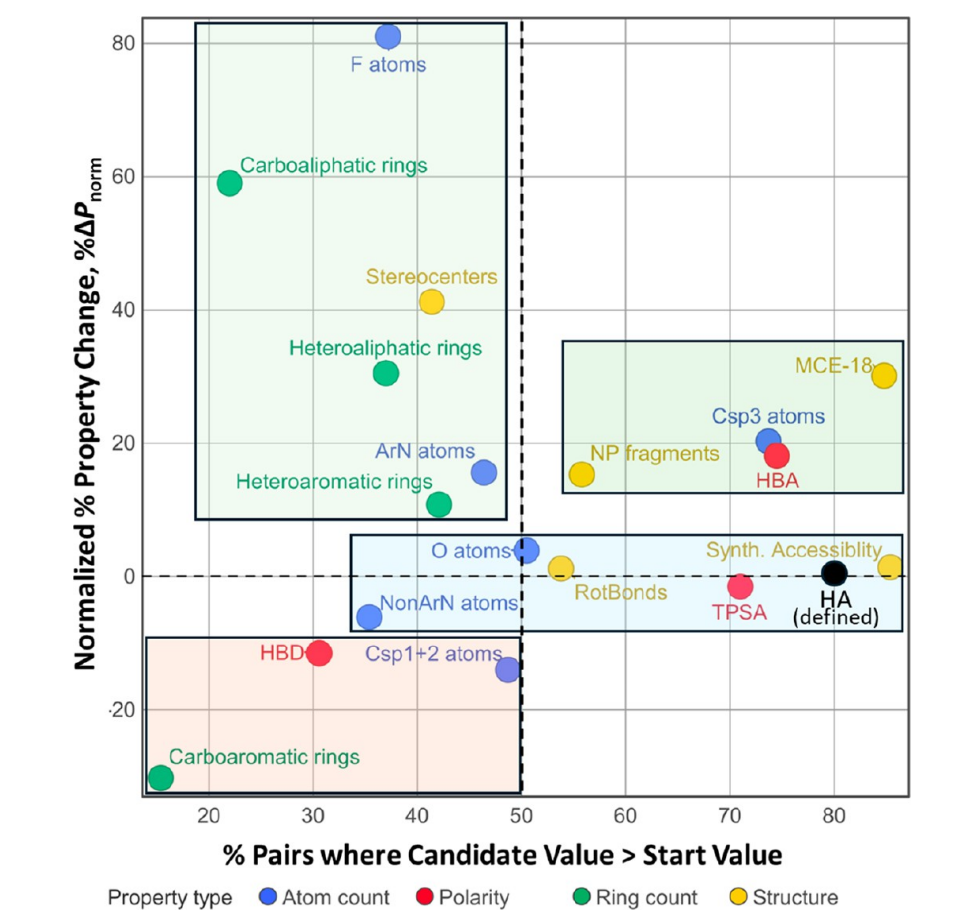
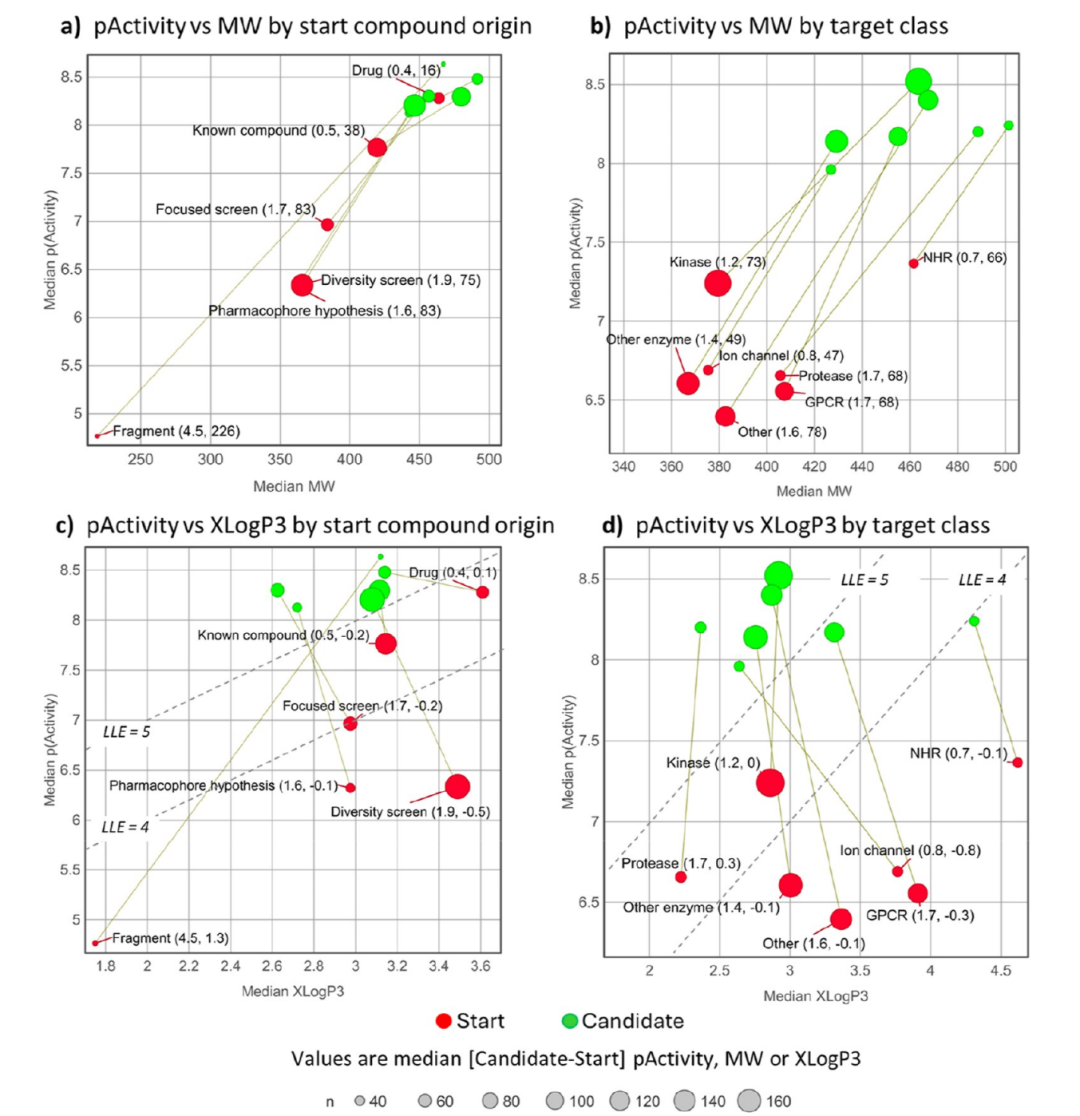
哪些性质真正推动了成功优化。MCE-18 复杂度、sp³ 碳、氢键受体和天然产物片段的增加超过了单纯分子变大的解释范围;碳芳香环、氢键供体和 sp²/sp¹ 碳则相对受到压制。
哪些性质真正推动了成功优化。MCE-18 复杂度、sp³ 碳、氢键受体和天然产物片段的增加超过了单纯分子变大的解释范围;碳芳香环、氢键供体和 sp²/sp¹ 碳则相对受到压制。
文章用 MCE-18 作为分子复杂度指标。这个指标强调脂肪环、螺环、手性中心和 sp³ 碳等特征。结果显示,候选药物的 MCE-18 平均从 52.7 增加到 80.7。更形象地说,许多成功分子不是从一张平面图纸变成更大的平面图纸,而是逐渐被折叠出更多空间结构。
当然,复杂不等于越复杂越好。文章也保留了另一类少数案例:约 9.2% 的优化从起点到候选物都停留在较低复杂度空间,主要由芳香环和非手性、非脂肪环取代基构成。这类路径如果可行,往往合成更直接、SAR 更清楚,也同样值得珍惜。问题在于,它不是大多数成功项目的常态。
第三层结论:成功优化并不喜欢从零重建,平均会保留七成起点骨架
药物化学项目往往看起来像不断重做分子,但数据给出的画面更保守。作者用最大公共子结构来衡量起点和候选物之间共享了多少骨架。结果显示,在整个数据集中,候选药物平均保留了起始化合物 70% 的重原子骨架;随机配对对照只有 34.4%。超过 50% 骨架保留的案例占 72%。还有 46 组,也就是 9.4% 的优化,完整保留了起始化合物的重原子框架,只是在外围做了小修小补。
这说明成功优化通常不是从一堆失败结构中完全推翻重来,而是在早期找到一个有价值的核心,然后围绕这个核心做连续、谨慎、可解释的改造。真正的难点并不是变化越多越好,而是尽早识别哪个部分值得保留,哪个部分必须替换。
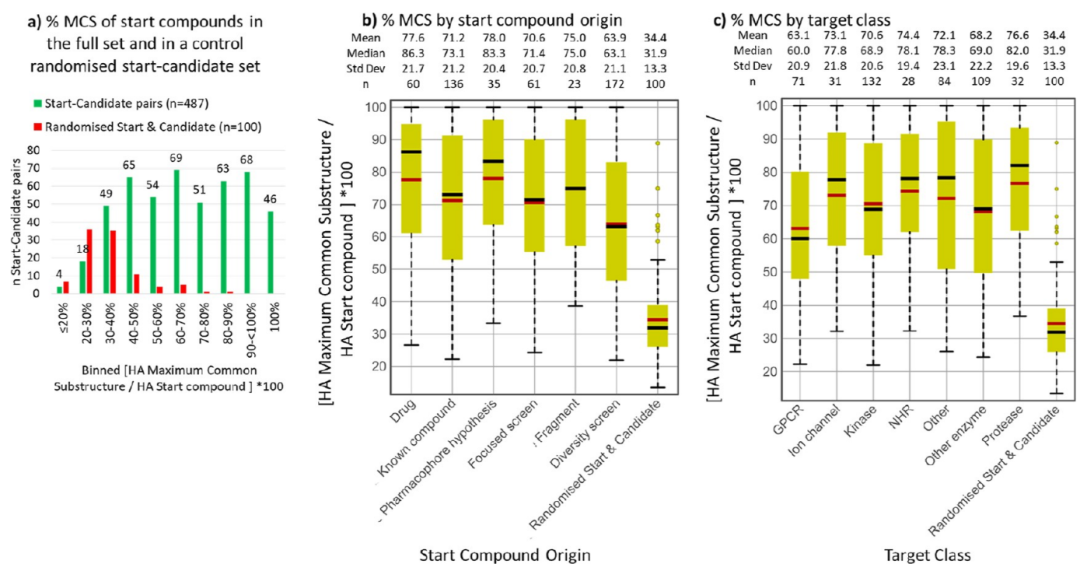
起始化合物与候选药物之间的骨架保留程度。成功优化平均保留约 70% 的起点重原子骨架,明显高于随机配对对照。
起始化合物与候选药物之间的骨架保留程度。成功优化平均保留约 70% 的起点重原子骨架,明显高于随机配对对照。
这也解释了为什么起始化合物质量如此关键。一个好的起点,不一定一开始活性很强,但它要允许药化团队沿着合理方向增长:可以增加分子量,可以提升复杂度,可以引入常见环系,同时还能控制脂溶性、氢键供体和总芳香环数。这样的起点才是真正有优化余地的起点。
先导样性需要重新理解:小而低脂溶仍然重要,但不能机械套公式
1999 年提出的经典先导样性标准是分子量小于 350、clogP 小于 3,且命中化合物不需要特别高活性。历史数据中,1995 年以前的先导化合物约 57.8% 符合这个分子量和脂溶性窗口。
但在这篇文章统计的当代筛选命中物中,只有 23% 同时满足分子量小于 350、clogP 小于 3;相反,40.5% 已经落在分子量大于 350、clogP 大于 3 的区域。原因并不难理解:靶点更难,结合位点更复杂,许多项目天然要求更大的分子去覆盖更大的空间。
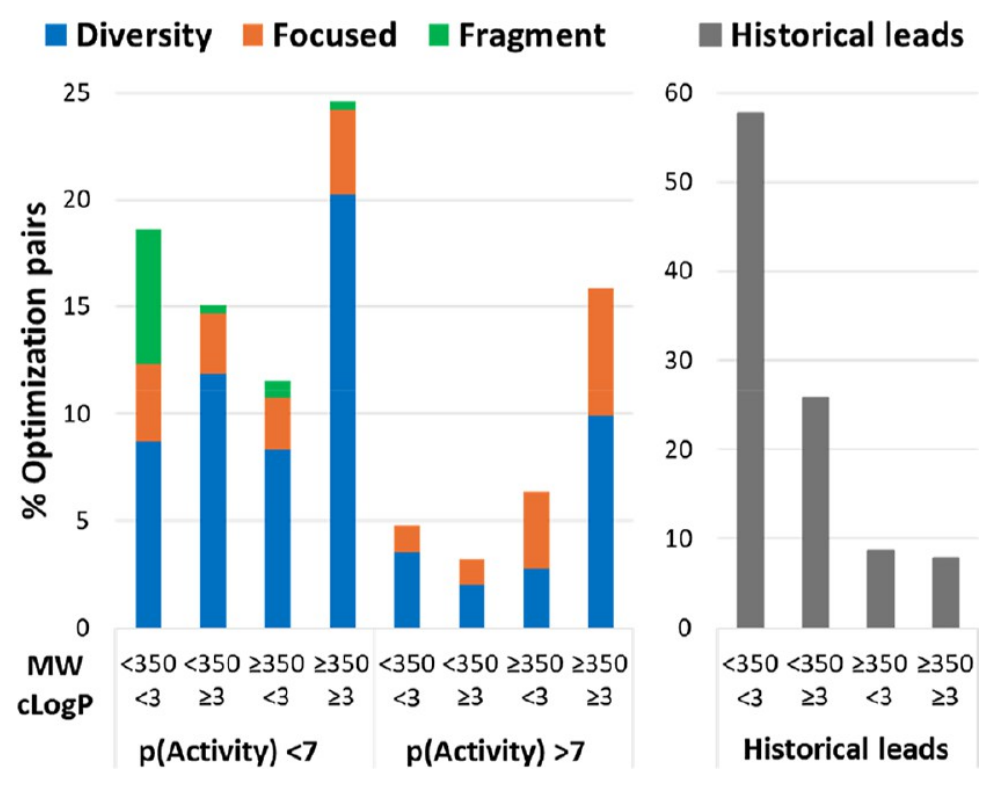
当代筛选命中物与历史先导化合物在分子量、脂溶性和活性空间中的差异。传统先导样性窗口在现代项目中仍有启发,但覆盖比例已明显下降。
当代筛选命中物与历史先导化合物在分子量、脂溶性和活性空间中的差异。传统先导样性窗口在现代项目中仍有启发,但覆盖比例已明显下降。
不过,传统先导样性并没有失效。文章按起始化合物分子量四分位数分析口服优化路径,发现最低分子量组,也就是 MW 小于 330 的起点,仍然呈现最经典的优化行为:平均分子量从 262 增至 402,活性从 5.9 增至 8.2,XLogP3 从 1.9 增至 2.5,HBA 从 3.3 增至 6.0,MCE-18 从 28 增至 61。它们的候选药物平均分子量也是最低的。
因此,对今天的药物发现来说,先导样性更适合作为起点可优化性的判断,而不是硬性筛选门槛。一个小而低脂溶的起点,如果没有清楚的生长方向,也未必好;一个稍大的起点,如果已经具备正确骨架、合适构象和可控脂溶性,也可能是高质量起点。真正重要的是:这个分子有没有能力在后续优化中提升活性,同时仍让脂溶性、氢键供体和芳香环负担留在可管理范围内。
五规则并没有消失,只是不能再被当成硬边界
在 445 个口服候选药物中,86.3% 符合五规则意义上的通过,即不违反或只违反一项规则。这个结果说明,传统口服小分子空间仍然是候选药物最主要的来源。
但细看违反项,会发现五规则的数字本身已经不那么整齐。分子量超过 500 的比例达到 30.6%,clogP 超过 5 的比例为 14%,O+N 超过 10 的比例为 8.1%,而 OH+NH 超过 5 的只有 0.9%。也就是说,分子量越界已经很常见,氢键供体反而被控制得非常严格。
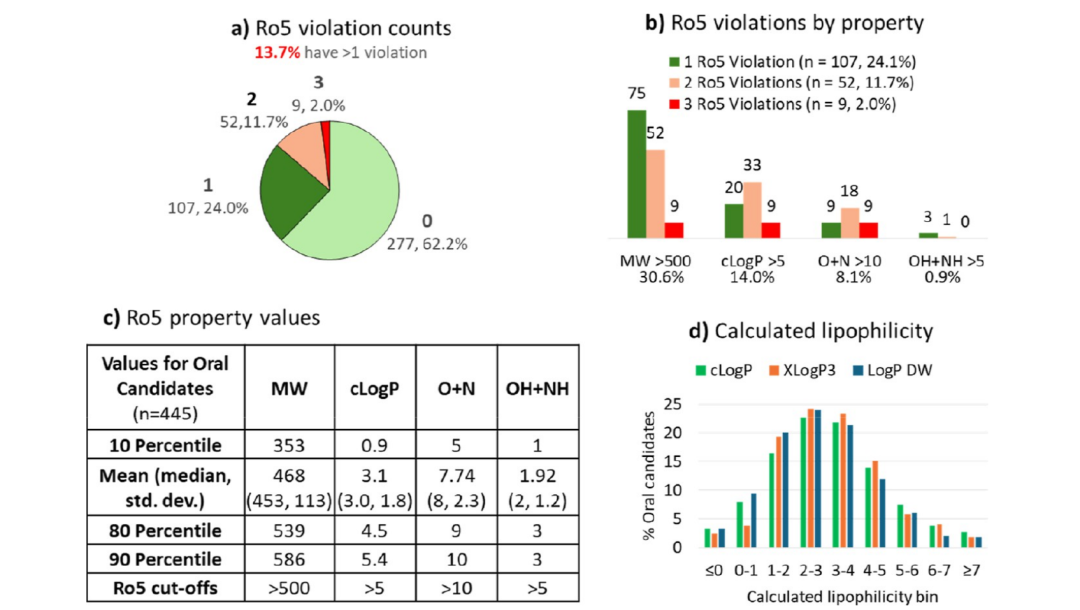
文章进一步采用扩展五规则和超越五规则的划分:口服候选物中分子量超过 500 的有 136 个,其中 102 个属于扩展五规则空间,34 个属于更典型的超越五规则空间。这提醒我们,今天的设计边界更像一条连续带,而不是一条绝对线。分子量可以上去,但脂溶性、暴露极性、氢键供体、构象柔性和溶剂暴露极性必须一起被考虑。
对于 PROTAC、宏环、分子胶这类新模态,文章中的案例数量还不多,不能把结论简单外推。但它已经指出一个方向:在高分子量空间里,传统的二维描述符不够用,溶剂暴露极性、分子内氢键、实验极性表面积、构象集合和变色龙性会越来越重要。这个判断对于传统五规则内分子同样有启发,因为口服吸收和组织分布从来不只由分子量决定。
案例里的现场感:KRAS 与 ATR 展示了知识如何被连续复用
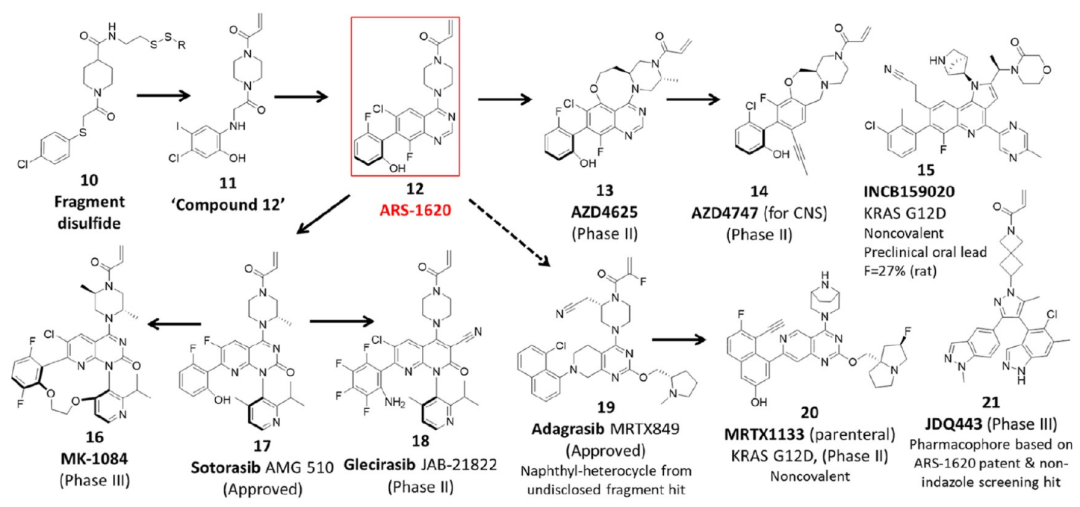
统计结果之外,文章还用大量案例展示了成功优化的真实路径。其中 KRAS 是很典型的一组。KRAS 曾长期被视为难以成药的靶点,G12C 共价抑制剂的突破来自片段系留策略发现 switch-II 口袋。ARS-1620 后来成为多个临床候选物的重要启发来源,进一步导向 sotorasib、adagrasib、JDQ443、MK-1084、glecirasib 等不同分子。这里看到的不是单个分子的胜利,而是一个早期结构发现如何被结构基础药物设计、骨架跃迁和共价策略不断放大。
ATR 激酶抑制剂则是另一种画面。一个含吗啉-嘧啶核心的聚焦筛选命中物,先后启发了 AZ20、ceralasertib、ART0380、AD1058,以及后续的 elimusertib、camonsertib、YY2201。多个候选物保留吗啉环,同时增加含氮杂芳环。这正好对应文章的总体趋势:常见环系被保留,杂芳环增加,分子复杂度上升,但脂溶性没有失控。
这些案例给人的启发是,成功优化很少是孤立灵感。它更像一条知识河流:早期结构、已知活性、晶体结构、失败毒性、代谢软点、竞争对手专利,都会成为下一轮分子设计的材料。药物化学家的工作不是在空白纸上画一个完美分子,而是在这些已有线索里判断哪一段值得继承,哪一段必须切掉。
对药物化学实践的启示:库设计和优化策略应更重视三维性
这篇文章对筛选库设计和命中物选择有很直接的提示。低分子量仍然重要,因为它提供增长空间;但低分子量本身不够,起点还应尽量具备可增长的三维复杂度、天然产物片段、小脂肪环和可控的杂原子模式。文章中常见且在优化中增加的小环包括吡啶、环丙烷、吡唑、吡咯烷、吗啉、嘧啶、吡喃和氧杂环丁烷。尤其氧杂环丁烷,在起始化合物中没有出现,却存在于 11 个候选药物中。
天然产物片段和伪天然产物也是一个值得关注的维度。候选药物中伪天然产物比例达到 66%,起始化合物为 46%;候选药物成为伪天然产物的概率约为起点的 2.3 倍。这里的意思不是天然产物样性本身带有神秘优势,而是药物化学实践越来越频繁地使用那些经过长期验证的小环和片段,把它们以非天然方式重新组合,得到既熟悉又有新构象的分子。
候选药物中出现频率较高并在优化中增加的环系。吡啶、环丙烷、吡唑、吡咯烷、吗啉、嘧啶、吡喃和氧杂环丁烷等常见小环,在成功优化中承担了重要结构角色。
对于正在做 hit-to-lead 或 lead optimization 的团队,这篇文章给出的实际判断可以压缩成几句话:分子可以长,但要知道往哪里长;活性可以用大小换,但脂溶性不能一路放行;苯环可以用,但不能让它主导整张分子图;氢键受体可以增加,氢键供体要谨慎;三维复杂度有价值,但复杂到难合成、难解释,也会反过来拖慢项目。
AI、速度与候选药物质量:快不是终点,认出好分子才是
文章最后讨论了人工智能和机器学习在药物发现中的位置。当前数据集中,AI/ML 主导发现策略的案例还很少。这并不代表 AI 不重要,而是因为公开候选药物文献存在时间滞后,许多数据反映的是 2010 年代中期前后的项目实践。
作者对 AI 的态度并不排斥,但很克制。虚拟筛选、蛋白结构预测、生成式分子设计、合成路线预测和体内外性质预测都会越来越有用,甚至可能改变命中发现的规模。但候选药物真正面对的是临床疗效和安全性,而这些不是单靠更快生成分子就能解决的问题。预测模型能不能可靠,取决于训练数据质量;分子能不能成为候选药物,取决于多参数平衡和项目判断。
文章提到一个很值得琢磨的现象:在多个企业回顾研究中,候选药物常常比团队意识到的更早出现。有些项目里,真正的候选物在候选系列中第几十个,甚至前几个化合物中已经被合成出来,只是后来经过更多验证才被正式确认。这个现象说明,优化不是无限追求更好分子,而是在复杂证据中识别已经足够好的分子。药化团队需要的不是永远多做一轮,而是知道什么时候该继续,什么时候该停下。
END:成功优化是一种有纪律的分子增长
这篇文章最终给出的不是一套神奇公式,而是一幅当代药物化学的平均画像。
成功优化中的候选药物通常更大、更复杂、活性更强,但平均脂溶性没有增加。分子增长主要来自 sp³ 碳、芳香氮、氧、氟、氢键受体和天然产物片段;碳芳香环减少,杂芳香环增加;氢键供体被严格控制。起点与候选物之间平均保留七成骨架,说明选择正确核心仍是成功的基础。五规则仍然有现实意义,但它更像经验坐标,而不是绝对边界。
真正值得带走的判断是:先导优化不是把分子做大,而是把分子长到合适的位置;不是把活性推高,而是在活性、脂溶性、极性、复杂度、代谢和安全性之间找到可以进入临床的平衡。
这也是药物化学长期迷人的地方。它需要数据,但不只是数据;需要模型,但不只是模型;需要规则,但又经常要知道何时越过规则。成功的候选药物,往往不是最漂亮的分子,而是在足够多限制条件下,仍然能工作的那个分子。
参考文献
What Happens in Successful Optimizations? A Survey of 2018–2024 Literature
Paul D. Leeson
Journal of Medicinal Chemistry 2026 69 (6), 6337-6395
DOI: 10.1021/acs.jmedchem.5c03171
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号