Nat. Commun. | 蛋白质结构中可拆分位点的准确预测

Nat. Commun. | 蛋白质结构中可拆分位点的准确预测

DrugAI
发布于 2026-06-24 13:26:50
发布于 2026-06-24 13:26:50
理解蛋白质结构架构,并预测其在发生深度重构时对结构扰动的耐受性,是功能蛋白质工程中的关键问题。研究人员提出了SplitSeek-Pro,这是一种深度学习模型,能够在折叠蛋白中以氨基酸残基分辨率评估“可拆分性”。这种能力对于环状置换、蛋白质拆分与重组等蛋白质工程任务十分重要。
SplitSeek-Pro将一级序列信息与三维结构特征结合起来,通过“大规模预训练—高质量微调”的两阶段训练流程,实现对每个残基位点可拆分概率的预测。研究人员在三种不同蛋白质上进行了实验验证,结果显示该模型相较于已有方法具有更强的预测能力。值得注意的是,SplitSeek-Pro还能识别出一些具有特征性的结构片段,这些片段表现为内聚、完整的结构单元,类似于超二级结构基序。研究人员认为,SplitSeek-Pro不仅是一个用于理性蛋白质工程的可靠工具,也为理解蛋白质折叠的基本结构模块提供了新的视角。为便于社区使用,研究人员还提供了自动化网络服务器。

蛋白质序列是决定蛋白质整体折叠和功能的最基本信息。若将蛋白质序列类比为一种“语言”,那么蛋白质的一级、二级、三级和四级结构可以被看作类似于文学中的词语、句子和段落。蛋白质工程在一定程度上就是对这种“蛋白质语言”进行重新表述或重写。基于结构的理性设计策略,例如环状置换、蛋白质拆分与重组、主链重连等,正是通过改变蛋白质主链连接方式来重新组织蛋白质功能。这些操作类似于语言中的倒装、重组或跨文本引用,但成功的前提是必须找到那些能够被安全切断或重新连接的位置,使蛋白质整体折叠和功能不被破坏。
过去,研究人员已经开发出多种计算方法,用于识别天然蛋白中发生过类似主链变化的实例,或预测可用于工程设计的环状置换位点和拆分位点。这些方法包括基于序列的工具、结构比对框架、传统机器学习服务器、能量景观和理化性质建模方法,以及高通量实验筛选平台。尽管这些方法推动了该领域发展,但在准确性、效率和泛化能力方面仍难以满足可靠识别主链可拆分位点的需求。解决这一问题不仅对环状置换、拆分蛋白设计、蛋白质传感器构建等工程任务具有实践价值,也有助于理解蛋白质折叠中的结构组织规律。
近年来,深度学习极大推动了蛋白质科学的发展。AlphaFold类模型在蛋白质结构预测方面取得突破,蛋白质语言模型能够提供丰富的序列表征,序列—结构联合模型和生成式蛋白质设计模型也已经广泛用于蛋白质设计。然而,将这些进展直接转化为通用的主链拆分位点预测仍面临一个重要瓶颈:高质量、大规模标注数据不足。直接的拆分—重组实验数据较少,而且容易受到片段溶解性、片段间亲和力、表达水平和体外重组条件等因素干扰。相比之下,环状置换数据更丰富,而环状置换和拆分重组共享一个共同前提,即蛋白质主链被打断后局部结构仍应具有足够稳定性。因此,环状置换数据可以作为训练拆分位点预测模型的有效代理数据。
基于这一思路,研究人员提出SplitSeek-Pro。该模型通过消息传递神经网络整合蛋白质序列和三维结构信息,预测折叠蛋白中每个残基位置对主链拆分的耐受程度。模型经过伪标签大规模预训练和实验数据微调,并在环状置换和拆分重组两类实验中得到验证,表现优于已有方法。
方法
研究人员将蛋白质表示为图结构,其中每个氨基酸残基作为一个节点,空间上相邻的残基对作为边。节点特征由蛋白质语言模型生成的序列表征和氨基酸理化性质共同初始化,边特征则由残基间原子距离信息编码,从而同时保留序列、理化和三维结构信息。模型主体采用编码器—解码器式消息传递神经网络,在蛋白质图中迭代更新节点和边信息,最后输出每个残基对应的可拆分概率。训练采用两阶段策略:第一阶段使用基于结构能量、序列保守性和溶剂可及表面积等特征生成的大规模伪标签数据进行预训练,以获得广泛的结构泛化能力;第二阶段使用环状置换数据库中经过筛选的实验验证数据进行微调,将已知环状置换位点作为正样本,同时从刚性二级结构区域中抽取负样本,并屏蔽不确定位置对训练损失的影响。模型性能通过计算测试、消融实验和湿实验验证综合评估,实验系统包括LCC、CHMO和NanoLuc,分别用于环状置换和拆分重组场景。
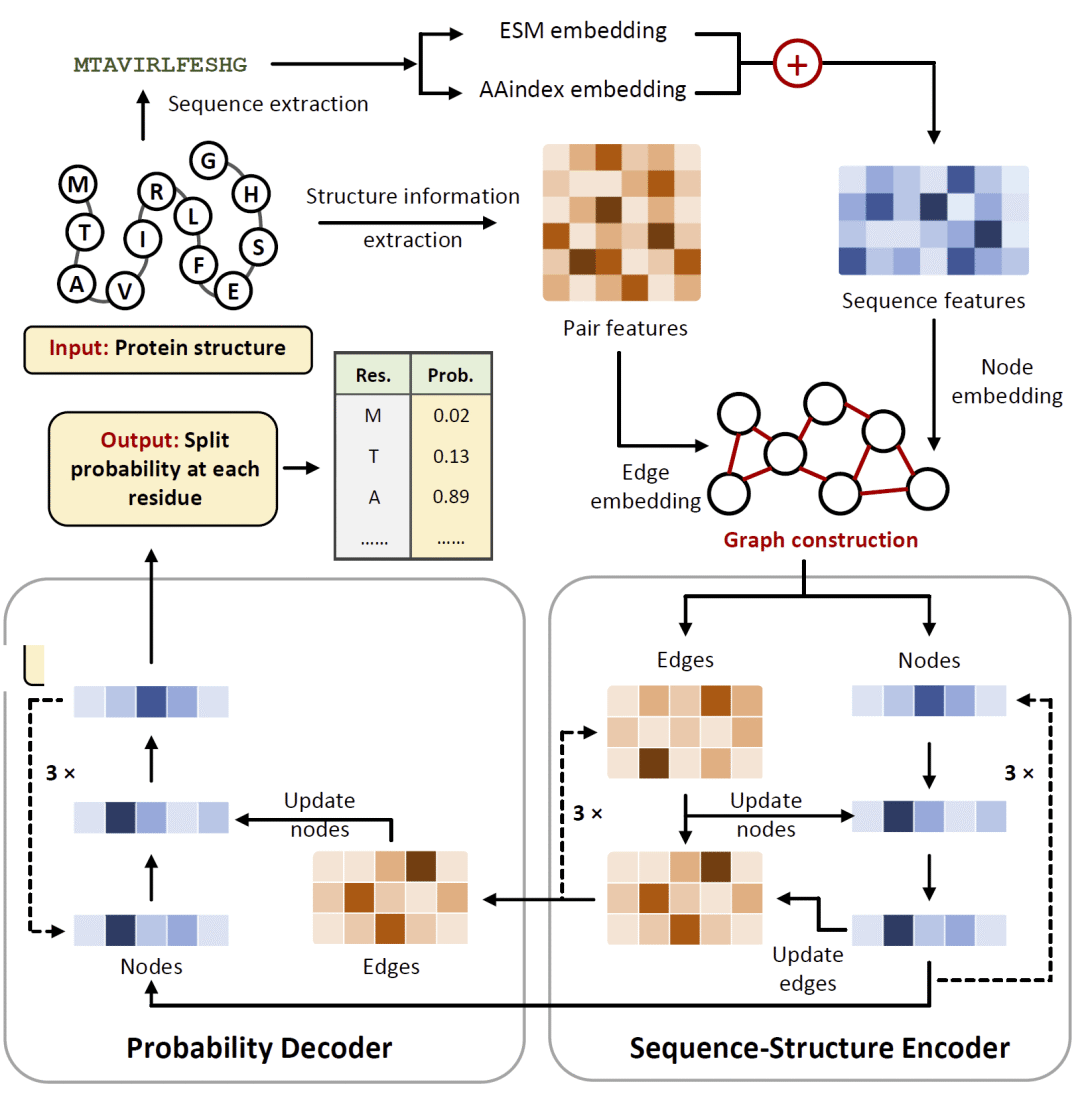
图1|SplitSeek-Pro模型框架与可拆分位点预测流程。
结果
深度学习模型设计
研究人员首先说明了SplitSeek-Pro的模型设计逻辑。理论上,蛋白质序列包含决定结构和功能的全部信息,因此从序列直接预测可拆分位点似乎是可行的。然而,缺少明确结构背景往往限制预测准确性。现代蛋白质语言模型虽然能够在一定程度上隐式捕获结构性质,但模型复杂、训练代价较高,也不一定最适合直接解决主链拆分位点预测问题。因此,研究人员认为,将序列信息与结构信息直接整合,是更有效的路径。
SplitSeek-Pro采用消息传递神经网络编码蛋白质。每个残基被视为图中的一个节点,节点特征由ESM-2生成的序列表征和AAindex中的理化性质共同组成。残基之间根据空间邻近关系建立连接,边特征则来自残基对之间特定原子的距离描述。通过这种方式,模型能够捕获蛋白质整体结构网络,而不仅仅依赖局部序列片段或二级结构类型。
模型由多层编码器和解码器组成,在编码过程中不断更新和整合节点与边的信息,在解码阶段输出每个残基的拆分概率。由于模型同时使用序列、理化性质和三维几何关系,因此能够避免单纯依赖二级结构类型作出判断。例如,模型不仅能识别许多环区位点,也能进一步区分哪些环区真正适合拆分,哪些环区虽然表面暴露但因功能或结构约束而不适合拆分。
数据集构建
蛋白质中某个位点是否“可拆分”,可以在不同实验语境下定义。理想情况下,如果从该位点拆分后形成的两个片段仍能独立保持一定结构,并通过非共价相互作用重新组装为功能蛋白,那么该位点可以被视为可拆分。相反,如果拆分导致蛋白质结构完整性丧失或功能无法恢复,该位点则被视为不可拆分。某些拆分蛋白系统需要借助外部相互作用模块提高两个片段的空间接近性,以促进重组。环状置换则是这一范式中的特殊情况:原始N端和C端通过连接肽相连,新的N端和C端出现在蛋白质内部位点附近,从而测试该位点对主链断裂和重新连接的耐受性。
由于直接的拆分蛋白实验数据有限,研究人员采用两阶段数据策略。预训练阶段使用伪标签数据。研究人员借助已有拆分位点预测思想,重新计算蛋白质每个位点的结构能量、二级结构、溶剂可及性和序列保守性等特征,并将这些特征组合为定量的可拆分概率标签。该过程为22,538个蛋白质生成了大规模预训练数据。
微调阶段使用环状置换数据库中的实验相关数据。研究人员筛选并优化后获得3,542对环状置换蛋白质对,从结构比对结果中提取拆分位点,并将其标记为高可拆分概率。为了平衡正负样本,研究人员从刚性二级结构区域中随机选择相同数量的残基作为负样本。对于没有明确标签的位置,则在训练中屏蔽,不参与反向传播。通过伪标签预训练与实验数据微调结合,SplitSeek-Pro同时获得了较好的泛化能力和面向真实工程场景的精确性。
模型性能
计算评估显示,预训练模型能够有效拟合低分位点,说明它已经学习到蛋白质能量、保守性和溶剂可及性等关键特征。然而,由于伪标签分布不均衡,预训练模型对高分可拆分位点的识别仍有限。经过实验数据微调后,模型区分高分和低分位点的能力显著提升。
研究人员还考察了图连接程度对模型性能的影响。当图中每个残基连接的邻居数量增加到一定程度后,模型对高分位点的预测能力得到改善,说明充分的结构上下文有助于识别可拆分位置。预训练也对最终性能有重要贡献,它提高了模型区分可拆分与不可拆分位点的能力。
对测试集中残基预测得分的分布分析显示,α螺旋和β折叠等刚性二级结构区域通常得分较低,而环区的得分分布更宽。这符合蛋白质工程中的常识:环区通常更适合作为环状置换或拆分重组的候选位置。然而,SplitSeek-Pro的重要优势在于,它并不是简单地把所有环区都判定为可拆分,而是能够在环区内部进一步区分不同位置的结构容忍度。研究人员也观察到一个局限:模型倾向于给蛋白质末端残基较低分数,而末端在理论上通常更容易被拆分。这一模式来自预训练阶段,微调后仍未完全改善,提示未来需要更多针对末端位点的训练数据。
消融实验显示,单独使用结构特征时,模型容易把许多环区都预测为高分,难以区分真正可拆分与不可拆分的环区;单独使用序列特征时,模型可以识别更多低分位点,但偶尔会把刚性二级结构中的残基误判为可拆分。完整模型同时使用序列和结构信息,表现最好,能够更清晰地区分高分和低分位置,并避免过度依赖二级结构类别。这说明,准确预测蛋白质主链可拆分性需要同时考虑序列演化约束、理化性质和三维结构背景。
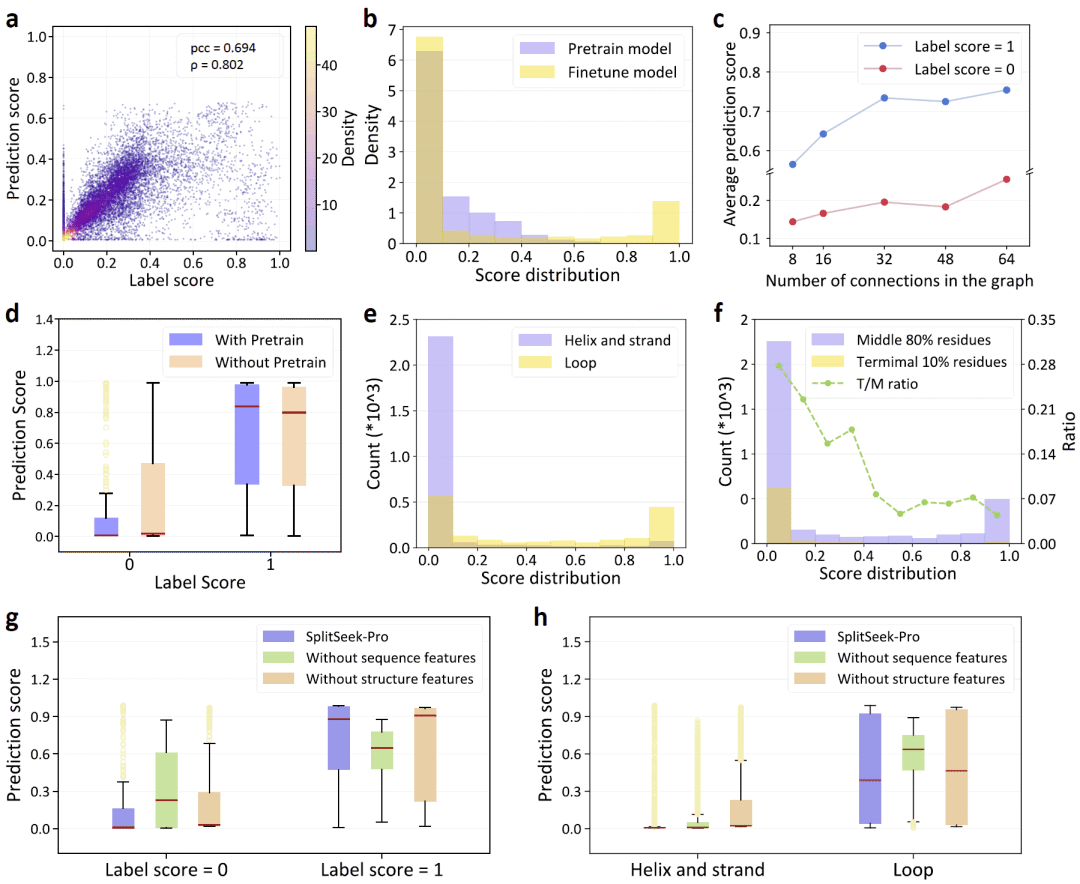
图2|SplitSeek-Pro的计算性能、训练效果和消融分析。
环状置换实验验证
研究人员首先用环状置换实验验证SplitSeek-Pro,因为该模型主要使用环状置换数据进行微调,而且环状置换所得蛋白仍为完整线性蛋白,便于通过表达、折叠和功能检测来评估结构完整性。研究人员选择了两个模型蛋白:LCC和CHMO。LCC是一种工程化聚对苯二甲酸乙二醇酯水解酶,CHMO是一种环己酮单加氧酶。二者具有不同折叠类型,并且与训练集中已有结构的序列和结构相似性较低,因此可用于检验模型泛化能力。二者的原始N端和C端在空间上较近,便于通过柔性连接肽连接,从而构建环状置换变体。
在LCC实验中,研究人员以预测得分0.50为阈值,将21个候选环状置换位点分为高分阳性组和低分阴性组。阳性组包含9个变体,全部位于环区;阴性组包含12个变体,其中部分位于环区,部分位于α螺旋或β折叠等刚性二级结构。实验结果显示,阳性组所有变体均可溶表达,只有一个表现出明显聚集;相比之下,阴性组中有多个变体无法观察到可溶表达,其余变体也更容易聚集。圆二色谱显示,可溶变体在不同程度上保留了二级结构,热稳定性测试也表明多数可溶构建体仍具有较高熔解温度,说明整体疏水核心未被严重破坏。
功能检测进一步支持模型预测。以小分子底物水解活性为指标,阳性组除一个明显聚集的变体外,基本保留水解活性;阴性组大多数变体失去活性,仅有一个位于α螺旋外周、远离活性位点的变体仍保持活性。PET降解实验也与小分子底物水解结果总体一致。研究人员指出,结构完整性和功能完整性并不总是完全一致:某些变体可能整体折叠仍存在,但因活性位点附近局部扰动而失去功能;也有部分变体虽然二级结构有所改变,但由于断裂位置远离活性中心,仍可保留催化活性。
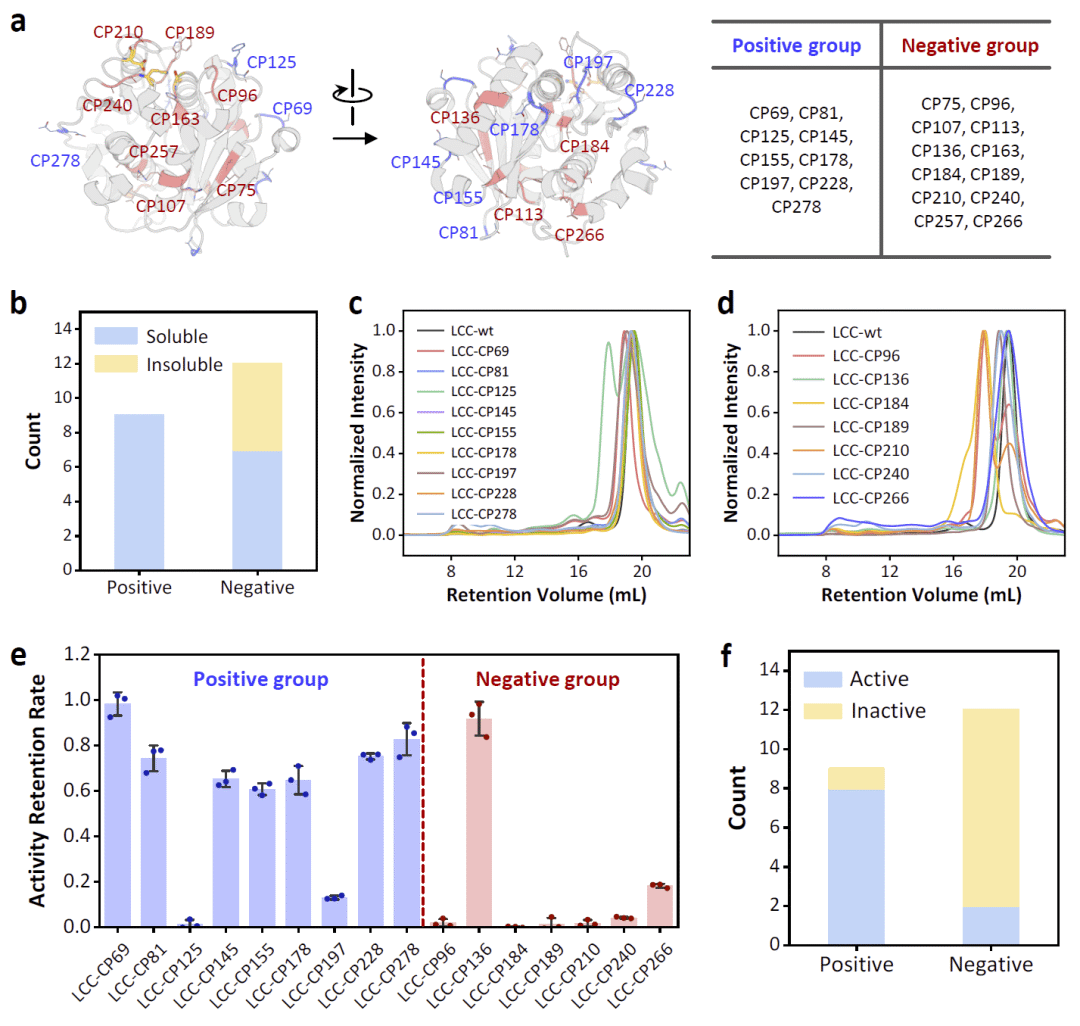
图3|LCC环状置换变体的实验验证。
在CHMO实验中,研究人员同样将候选位点分为阳性组和阴性组。阳性组10个变体中有8个可溶表达,其中只有一个可溶变体出现明显聚集。阴性组18个变体中有8个无法可溶表达,其余10个也在体积排阻色谱中显示明显寡聚化倾向。圆二色谱表明可溶变体总体保留了二级结构特征,但其中两个变体无法检测到明确熔解温度,提示结构不稳定。
CHMO的功能依赖FAD辅因子结合。研究人员检测了变体是否具有FAD吸收峰,并进一步使用环己酮氧化反应监测NADPH消耗来评估酶活性。阳性组变体通常表现出一定活性,但一个无法结合FAD的变体例外。部分阴性组变体虽然能够结合FAD,但催化活性很低。结构分析显示,这些位点靠近底物结合口袋,说明拆分虽然未破坏辅因子结合,却可能严重扰动底物口袋,从而导致功能几乎完全丧失。
总体来看,LCC和CHMO实验验证了SplitSeek-Pro对环状置换位点的预测能力。虽然所有环状置换变体相比野生型都有不同程度活性下降,但许多高分位点变体仍保持较好结构和功能。研究人员认为,模型在环区位点预测上表现尤其强,但对刚性二级结构内部可拆分位点和末端附近位点仍有改进空间。
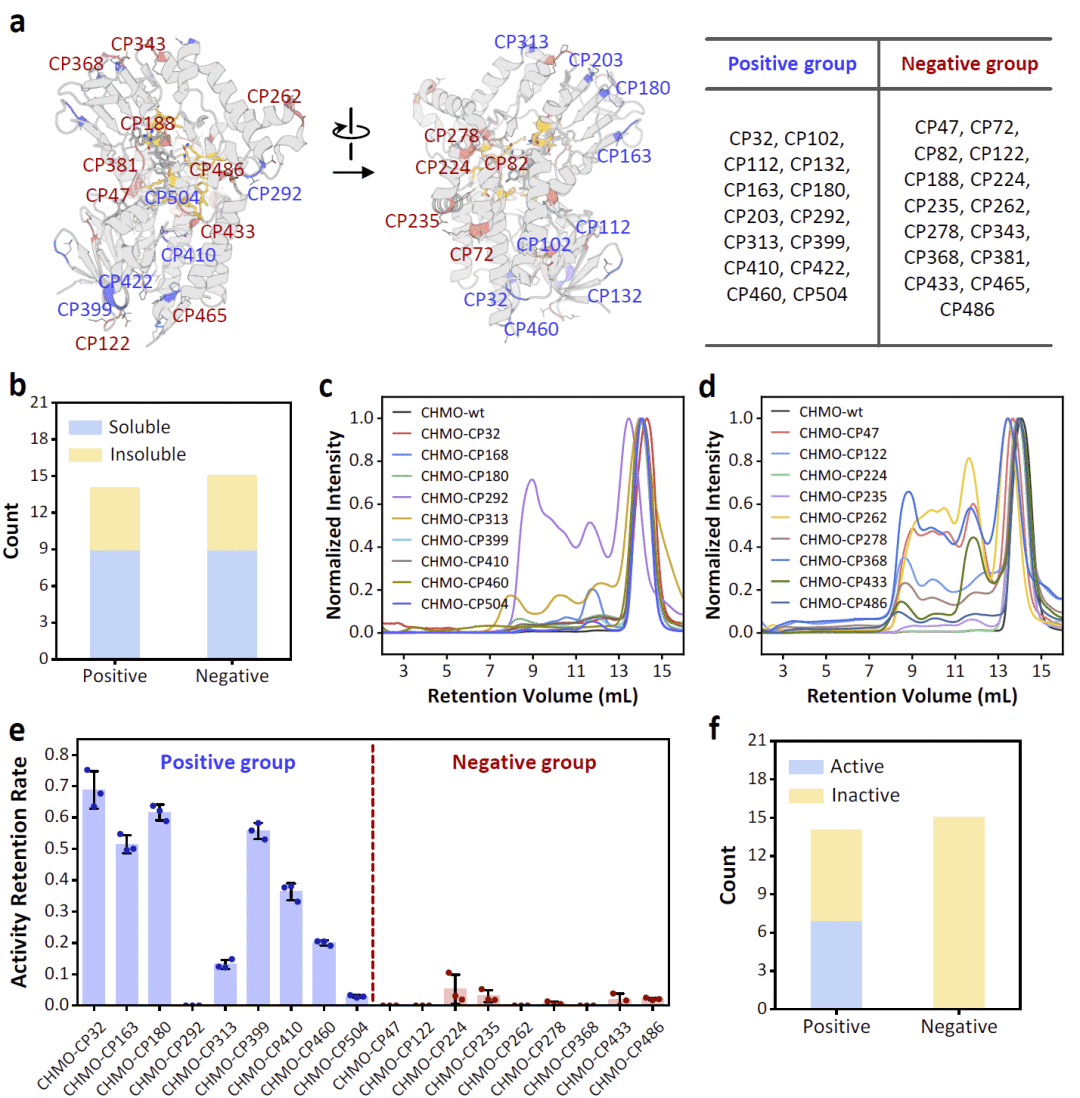
图4|CHMO环状置换变体的实验验证。
拆分与重组实验验证
在环状置换验证之后,研究人员进一步评估SplitSeek-Pro能否指导拆分蛋白对的设计。与环状置换相比,拆分重组更加困难,因为两个片段不再由共价连接肽约束,可能出现片段聚集、不可溶表达、错误折叠或动力学陷阱等问题。研究人员选择NanoLuc作为模型系统,因为它体积小、发光强,且拆分NanoLuc对是研究蛋白质相互作用的常用工具。NanoLuc与训练集序列相似性较低,也适合作为泛化测试对象。
已有研究报道过NanoLuc中若干功能性和非功能性拆分位点,这些结果与SplitSeek-Pro预测总体一致。基于模型建议,研究人员选择了四个新位点进行实验测试,其中包括两个高分位点、一个低分位点,以及一个因邻近残基得分较高而被认为有潜力的位点。为了评估片段在自发重组和强制接近条件下的表现,研究人员设计了两套实验系统:一套仅依赖片段自身相互作用,另一套使用SpyTag/SpyCatcher系统将两个片段拉近,以促进重组。为避免干扰活性位点,SpyTag和SpyCatcher被融合在远离催化口袋的片段末端,同时还引入GFP融合以改善大片段溶解性。
实验结果显示,除一个构建体外,多数片段均可溶表达,但表达量较低。发光活性检测表明,预测高分的两个拆分对能够产生可检测发光,说明它们可以成功自发重组。在SpyTag/SpyCatcher辅助下,这些片段重组更快,活性在混合后短时间内达到峰值,随后进入平台期并略有下降。
然而,并非所有高分位点都能成功重组。一个高分位点对应的拆分对即使在SpyTag/SpyCatcher辅助下也几乎没有发光。研究人员认为,这反映了环状置换与拆分重组之间的热力学差异。在环状置换中,共价连接肽类似于分子内伴侣,可以帮助两个片段保持足够接近,只要片段具有一定折叠稳定性,蛋白仍可能恢复功能。而拆分重组必须克服更高的热力学障碍,尤其是两个独立片段结合时的平移熵惩罚,因此需要片段间具有较强亲和力或较高的单片段稳定性。SplitSeek-Pro擅长识别那些对全局结构扰动较小的拆分位点,但对孤立片段能否自发高效重组的预测仍有限。尽管如此,模型仍能为可行拆分重组位点提供具有实用价值的预测。
SplitSeek-Pro性能评估
为了系统评价SplitSeek-Pro,研究人员将其与CPred和Int&in等已有工具进行比较。首先,研究人员使用来自LCC、CHMO和NanoLuc实验的54个统一实验样本,并以活性保留是否达到一定阈值来划分真实可行或不可行位点。结果显示,SplitSeek-Pro在识别真正可行位点方面与CPred相当,但在排除假阳性方面明显更好,尤其能更有效拒绝实际不可用的拆分位点。Int&in也具有一定区分能力,但更容易将不可行位点误判为可行。SplitSeek-Pro的优势在环区尤为突出,因为大多数候选拆分位点本身就位于环区,而模型能进一步区分哪些环区具有真实可拆分性。
随后,研究人员整合更多文献数据,构建了一个包含526个精炼数据点的独立测试集,用于评估环状置换和拆分重组位点预测能力。在准确率、曲线下面积、F1分数和Matthews相关系数等指标上,SplitSeek-Pro均优于CPred和Int&in。研究人员还与SPELL进行了代表性子集比较,结果显示两者在推荐核心位点方面具有一定重叠,但SplitSeek-Pro能够识别更多在实际实验中保持功能活性的潜在拆分位点。此外,研究人员测试了AlphaFold2和ESMFold等结构预测工具是否能直接用于该任务,发现它们无法有效区分可行环状置换位点。这说明,结构预测本身并不等同于可拆分性预测,仍需要专门模型。
研究人员也分析了SplitSeek-Pro的假阳性和假阴性。假阳性常出现在表面暴露残基上,这些残基虽然溶剂可及且不属于疏水核心,但对结构稳定性或功能仍十分关键。某些高分位点即使整体结构可能仍能保持,也可能因远距离“幽灵效应”影响催化功能。为降低假阳性,研究人员建议,当至少连续两到三个残基都具有较高预测分数时,该区域才更可靠地作为可拆分候选。假阴性则常源于模型难以准确判断刚性二级结构中的某些特殊可拆分位点。由于训练集中这类例子较少,模型倾向于将α螺旋和β折叠内部位点判定为不可拆分。未来更丰富、更高质量的数据有望改善这一问题。
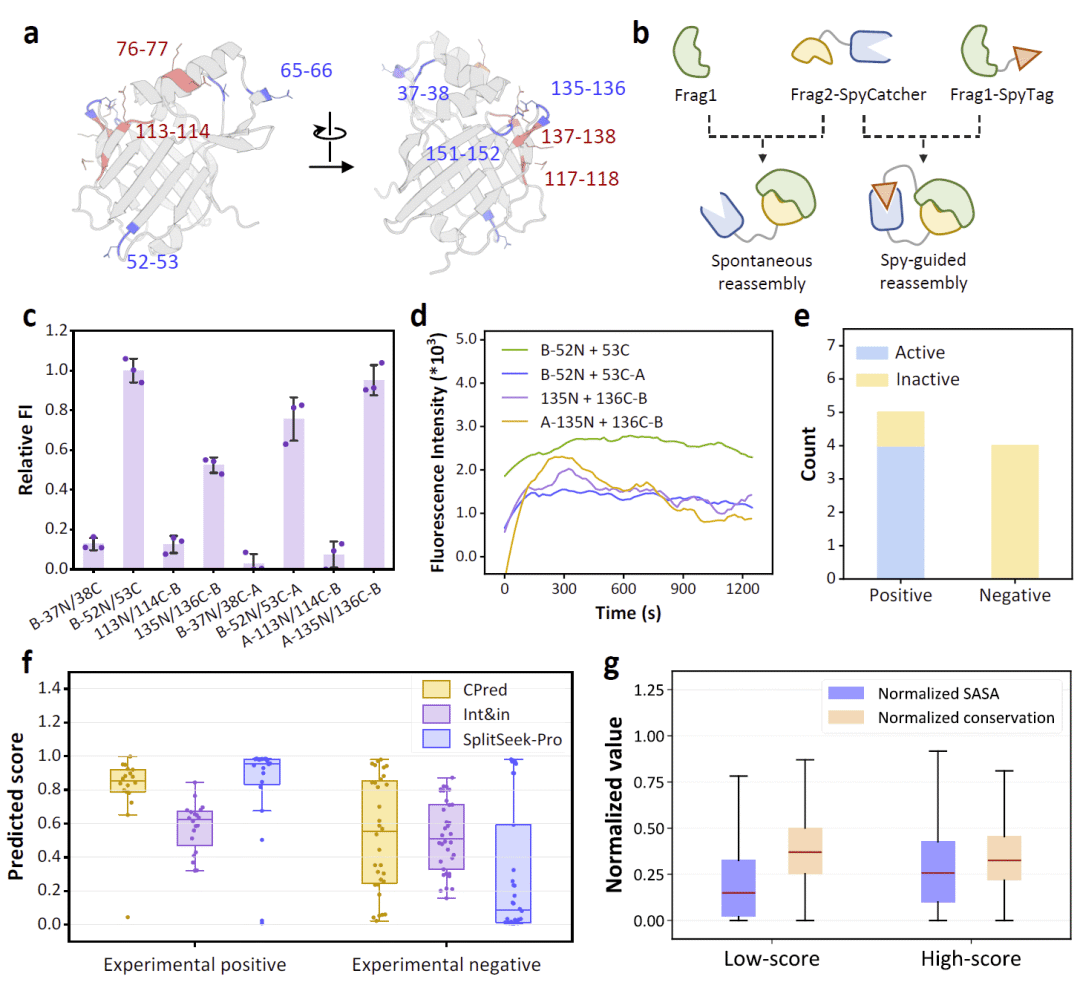
图5|拆分NanoLuc蛋白对表征与SplitSeek-Pro性能评估。
SplitSeek-Pro的结构生物学意义
SplitSeek-Pro的高准确性使研究人员能够进一步系统分析可拆分位点及其对应结构片段的序列和结构特征。研究人员利用SCOPe蛋白结构分类数据库,在控制序列相似性的基础上分析模型预测结果,并以0.50作为区分高分和低分位点的阈值。结果显示,低分位点通常具有较低溶剂可及性和较高序列保守性,而高分位点往往更暴露、更不保守。这符合蛋白质工程直觉:埋藏在疏水核心或受强进化约束的残基通常更不耐受主链拆分。
有趣的是,在LCC和CHMO中,SplitSeek-Pro会给活性位点附近的一些环区残基较低分数。尽管这些位点属于环区,但它们可能具有重要功能约束。这说明,模型并不是单纯依赖结构暴露程度或二级结构类型,而可能通过蛋白质语言模型表征捕获了序列保守性和功能约束信息,从而识别出不应被拆分的功能关键区域。
研究人员进一步使用SplitSeek-Pro高分共识拆分位点将蛋白质切分为结构片段,并发现这些片段并非随机分割结果,而常常对应具有协同行为的超二级结构模块。按所含二级结构元件数量分类后,研究人员观察到许多多元结构基序,且随二级结构元件数量增加,片段频率逐渐下降。对于包含两个二级结构元件的片段,主要出现β-β角、含α螺旋的角状结构以及较少见的α-β排列。其中,β-α角状结构占比较高,提示其在三级结构形成中可能具有重要作用。α-α角状结构相对少见,这一趋势在包含三个或四个二级结构元件的片段中也存在,提示α螺旋可能具有更强结构自主性,而β折叠更倾向于与其他结构元件协同形成稳定单元。
对于包含更多二级结构元件的片段,研究人员识别出全β基序、α-β交替基序等已知超二级结构模式。这些SplitSeek-Pro片段处于普通二级结构和完整三级折叠之间,构成了一类中尺度结构单元。研究人员还将这些片段与已有实验解析的蛋白质折叠路径进行比较,发现一些早期折叠单元往往位于SplitSeek-Pro片段内部,并且片段内部组成元件之间的拆分分数较低。这说明这些片段可能作为协同折叠模块,在三级结构形成中发挥关键作用。
SplitSeek-Pro片段也与既有蛋白质结构基序数据库存在呼应,但二者定义方式不同。传统结构基序方法多基于主链几何,而SplitSeek-Pro同时利用序列和结构信息。因此,SplitSeek-Pro识别出的片段通常更紧凑,内部二级结构元件相关性更强;也有一些松散延展的片段可能充当刚性结构块之间的连接器,类似折叠结构域中的环区。这些片段在大量天然蛋白结构中反复出现,可能代表早期折叠片段或进化中保留下来的祖先结构模板。研究人员还将SplitSeek-Pro高分区域与多结构域蛋白中的结构域边界进行比较,发现二者高度一致,说明模型能够识别相对独立的结构片段或结构域之间的可拆分连接区。
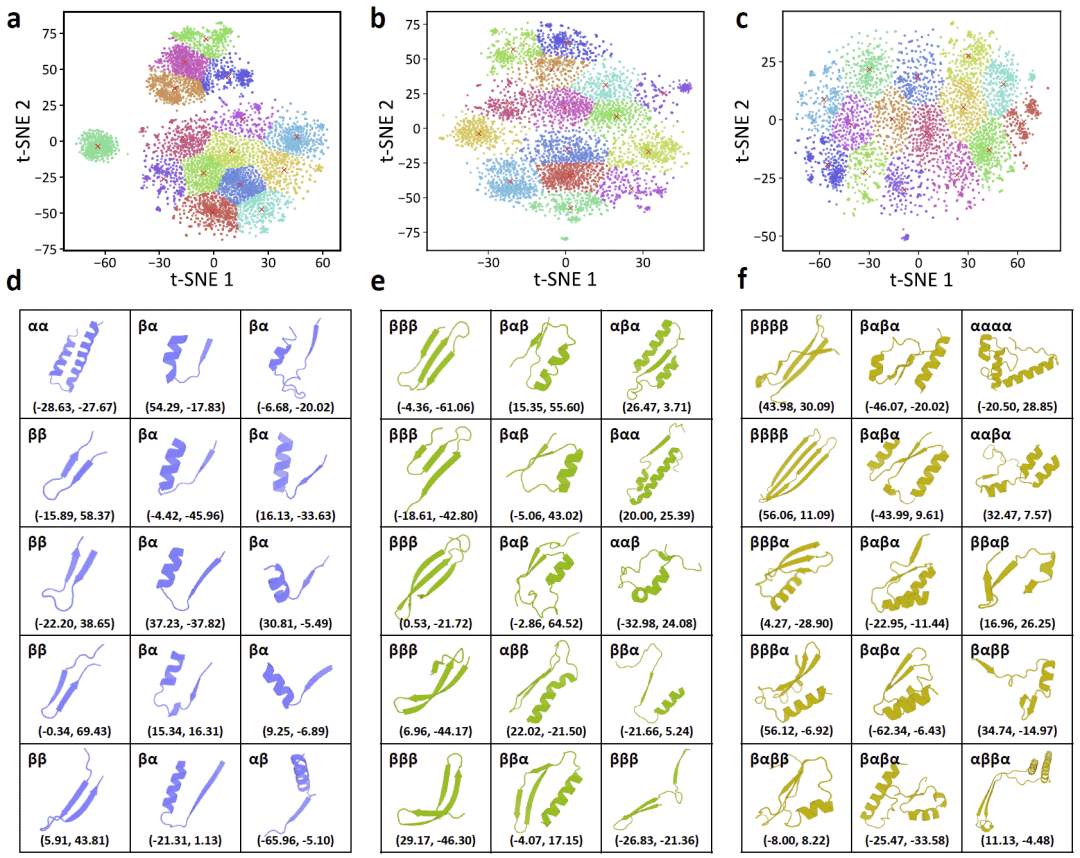
图6|SplitSeek-Pro片段聚类得到的结构基序可视化。
讨论
研究人员提出了SplitSeek-Pro,这是一个能够整合序列和结构特征、准确预测折叠蛋白中可拆分位点的深度学习框架。通过LCC和CHMO的环状置换实验,以及NanoLuc的拆分重组实验,研究人员证明该模型能够较可靠地区分可行和不可行的主链拆分位点,并在多个指标上优于CPred、Int&in等已有工具。SplitSeek-Pro的价值不仅在于为蛋白质工程提供实用预测工具,也在于它能够揭示蛋白质折叠中的中尺度结构模块,即研究人员所称的SplitSeek片段。这些片段类似超二级结构基序,往往表现为协同折叠和结构完整性维持的基本单元。
从工程角度看,SplitSeek-Pro可用于指导环状置换、拆分蛋白设计、蛋白质重连、蛋白质传感器构建以及功能模块重排等任务。与传统经验方法相比,它能够在全蛋白范围内给出残基分辨率预测,从而显著缩小实验筛选空间。尤其是在环区众多、人工判断困难的蛋白质中,模型可以进一步区分真正具有结构容忍度的环区和虽然暴露但功能上受限的环区。
不过,研究人员也承认模型仍存在边界。首先,对于未知结构蛋白,SplitSeek-Pro可以使用AlphaFold预测结构作为输入,但对于本征无序蛋白,模型性能会受到限制,因为这类蛋白缺乏稳定折叠结构,而SplitSeek-Pro主要面向折叠蛋白。其次,训练数据仍有限,尤其缺乏末端残基和刚性二级结构内部可拆分位点的高质量实验样本。因此,当前模型更适合进行二分类式可行性判断,而不是精确预测拆分后结构变化程度或功能保留比例。
模型的假阳性和假阴性也提示了未来改进方向。某些表面暴露位点虽然模型评分较高,但实际对结构稳定或催化功能至关重要;某些刚性二级结构内部位点虽然模型评分较低,却在实验中仍可成功拆分。这说明主链拆分耐受性受到多因素共同影响,包括局部结构、全局折叠核心、活性位点距离、进化保守性、片段溶解性和片段间重组能力。未来模型需要更多“干实验—湿实验”闭环数据,通过迭代设计、实验验证和任务特异性微调不断提升。
研究人员认为,现有结构预测工具本身无法判断蛋白质可拆分性,因为折叠结构是否可被准确预测,并不等同于主链在某处断裂后仍能保持结构和功能。SplitSeek-Pro正是面向这一特定工程问题建立的专用模型。未来,如果能够结合更多实验数据、回归式功能保持标签、片段稳定性信息和重组动力学信息,SplitSeek-Pro有望从当前的可拆分位点分类工具,进一步发展为能够定量预测结构扰动和功能保留的蛋白质工程设计平台。
整理 | DrugOne团队
参考资料
Wang, YX., Jiang, F., Xu, T. et al. SplitSeek-Pro: accurate prediction of splittable sites on protein structures. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-74059-z

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-20,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号