Nat Chem Biol | 把针剂变成药片

Nat Chem Biol | 把针剂变成药片

MindDance
发布于 2026-06-29 14:17:38
发布于 2026-06-29 14:17:38
从 8448 个环肽里,造出一个能口服的凝血酶抑制剂
很多最该被攻克的疾病蛋白,至今没有一颗能吞下去的药。有些蛋白表面平坦光滑,小分子找不到能嵌进去的口袋;有些虽有口袋,却很难做到只认它、不认它的同族兄弟。能解决这类难题的,往往是抗体这样的大分子生物药——可它们几乎都得靠注射进入体内。
夹在小分子和生物药之间的环肽,本来是个诱人的中间地带:足够小,又能像大分子一样咬住难缠的靶点。自然界也给过证明,环孢素 A、去氨加压素、一种生长抑素类似物,都是来自天然、能口服的肽类药物。但它们是少数幸运儿。绝大多数天然或人工设计的肽,要么个头大、表面太亲水,要么扛不住消化道里的蛋白酶,最后只能做成针剂。

这项来自洛桑联邦理工学院(EPFL)Christian Heinis 团队、发表在 Nature Chemical Biology 上的工作,想回答一个一直悬而未决的问题:能不能从零开始,针对一个具体靶点,造出既能精准结合、又能口服吸收的小环肽。
〔名词小注:口服生物利用度(%F)〕一颗药吃下去,真正进入血液循环、能去发挥作用的比例,就是口服生物利用度,记作 %F。把同一种药分别口服和静脉注射,比较两条血药浓度曲线下的面积,就能算出来。静脉注射默认是 100%;口服能有百分之十几,对一个肽来说已经相当难得。
三道关,彼此较劲
口服一个肽,难在它要同时闯过三道关,而这三道关彼此较劲。
第一关是消化道里的蛋白酶。胃里的胃蛋白酶、肠道里的胰酶,本职工作就是把蛋白和肽剪碎成氨基酸。第二关是肠壁:分子必须穿过肠上皮细胞的脂质膜才能被吸收,而肽往往太大、表面带的极性基团太多,像一团亲水的线球,过不去那层油膜。第三关是肝脏:被吸收进门静脉的分子,第一站就是肝,那里挤满了代谢酶,会给分子打上各种修饰、加速它被清除,这叫首过代谢。
人们早就总结出能口服的肽大致长什么样:分子量在 700 道尔顿以下、极性表面积小于 200 平方埃、氢键供体不超过五个。但这并非铁律——环孢素 A 重达 1,203 道尔顿,靠大量 N-甲基化和能蜷缩起来藏住极性表面的构象,照样能口服。
〔名词小注:五规则(rule of five)〕一组判断小分子能否口服吸收的经验阈值,由 Lipinski 提出,因为几个关键数字都与 5 有关(氢键供体不超过 5、受体不超过 10、分子量不超过 500 等)而得名。环肽个头比典型小分子大,几乎都越出了这个范围——它们落在所谓五规则之外的空间,是介于经典小分子和生物药之间的一片灰色地带。
真正的难处不在画出这张理想画像,而在造出符合画像、又恰好能结合某个具体靶点的肽。要找到这样的分子,得在海量候选里筛。过去靠噬菌体展示或 mRNA 展示来建大库,但这类生物展示库的化学多样性有限,大多只能用天然氨基酸,很难做出又短又强的结合肽。Heinis 团队此前用噬菌体展示做过抗消化道蛋白酶的环肽,即便最小的也还有九个氨基酸、1,134 道尔顿,太大太亲水,在小鼠体内的口服生物利用度只有 0.2%。
从二硫键,换到硫醚键
Heinis 团队此前的解法,是在微孔板里用声波分液的方式,以纳摩尔的微量规模把环肽一个个组合合成出来,粗产物不经纯化直接拿去筛——他们曾这样造出两万个环肽,并找到纳摩尔级的结合分子。但那批环肽是用二硫键环化的,而二硫键有个致命短板:它在还原环境里会被打开。
这正是口服的拦路虎。分子穿过细胞时,细胞内部是还原性的环境,二硫键会被还原、环被打开,肽就散了架。于是这套高效的合成筛选方法,一直没法用来做口服肽。这项工作的关键一步,就是把环化用的化学键从二硫键换成硫醚键。
〔打个比方〕二硫键像一对用磁力吸在一起的搭扣——做的时候一碰就合上,很省事,可一旦环境合适(比如细胞内的还原环境),它也会被轻轻一掰就开。硫醚键则像把这对搭扣焊死了:同样能把环扣起来,却不怕被还原性环境拆开。代价是焊点(那个硫原子)有自己的软肋,后面会提到。
硫醚环化还有一个对建库极其友好的优点:这个反应又干净又高效。两端带巯基的线性肽,和一个带两个反应位点的连接子一拌,几乎定量地就环化好了。正因为干净,粗产物里杂质少,才敢不纯化直接拿去筛——这是能上量、做大库的前提。
不过硫醚键的软肋也得先摸清:硫原子容易被氧化,而氧化主要来自肝脏里 CYP450 家族的酶,也就是首过代谢那一关。团队先合成了十个结构各异的双硫醚环肽 1–10,放进大鼠肝微粒体(从大鼠肝细胞分离出、富集了代谢酶的组分,常用来预测肝脏代谢清除的快慢)里,看它们扛不扛得住。结果差别很大:最不稳定的肽 1 半衰期只有约 11 分钟,最稳定的肽 2 长达 133 分钟;被氧化的肽会多出 16 或 32 的质量,对应加上一个或两个氧原子。拿两个已上市的口服药维拉帕米和地尔硫卓做标尺,十个环肽里有九个和它们相当或更稳,其中最稳的四个清除率还要慢四倍以上。
〔Aha 时刻〕硫醚键虽然会被氧化,但只要选对序列,它的稳定性足以达到、甚至超过已经在临床上口服使用的药。也就是说,把二硫键换成硫醚键,既保住了建大库所需的高效环化,又跨过了穿不过还原环境这道坎——剩下的氧化问题,可以留到后面慢慢治。
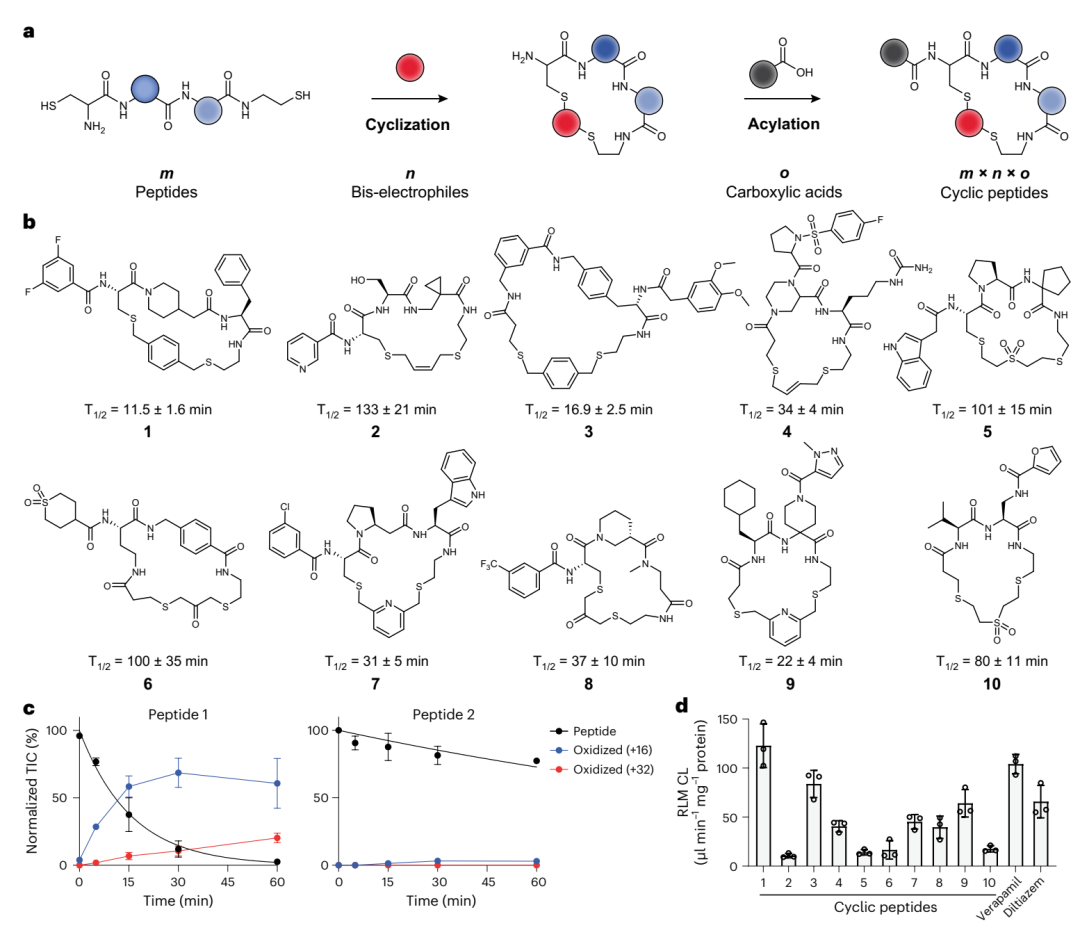
双硫醚环肽的合成思路与代谢稳定性。a 是整套建库策略的总览:两端带巯基的线性肽先用双亲电连接子环化,再在外围氨基上接一个羧酸,m 种肽 × n 种连接子 × o 种羧酸,组合出庞大的环肽库。b 给出十个随机环肽 1–10 的结构和各自在大鼠肝微粒体里的半衰期,可直观看到稳定性差异之大。d 把十个肽与两个口服药的清除率排在一起,看哪些比药更稳。
双硫醚环肽的合成思路与代谢稳定性。a 是整套建库策略的总览:两端带巯基的线性肽先用双亲电连接子环化,再在外围氨基上接一个羧酸,m 种肽 × n 种连接子 × o 种羧酸,组合出庞大的环肽库。b 给出十个随机环肽 1–10 的结构和各自在大鼠肝微粒体里的半衰期,可直观看到稳定性差异之大。d 把十个肽与两个口服药的清除率排在一起,看哪些比药更稳。
不纯化,直接筛
要把这套思路变成能上量的合成,核心是把两步反应串起来、且中间不做纯化:先做硫醚环化,紧接着在环肽外围的氨基上接羧酸(酰化)。两步都只需往微孔板里按顺序加试剂。
这两个反应过去各自都用过,却没人把它们接在一起。团队先用八个两端带巯基、还带一个氨基的随机肽(P1–P8)探路。这些肽在 96 孔板里用半胱胺树脂合成,靠乙醚沉淀就能拿到不错的纯度,八个肽平均纯度达到 93% ,全程不用色谱纯化。
接着测环化:把八个肽分别与四个连接子(L1–L4)两两组合,共 32 个反应。为了让分子内成环压过分子间副反应,反应在低浓度(1 毫摩尔)、大体积(1 毫升)下进行,连接子用两倍量。32 个反应的环化效率平均 85% ,最主要的副产物是二硫键环化的肽,大多数反应里不到 1%,最多也只有 15%。再测酰化:把环化好的肽与八个羧酸(A1–A8)组合,在 384 孔板里以 500 皮摩尔的微量、50 纳升的反应体积进行,室温 8 小时,结果是几乎定量的酰化。
〔打个比方〕这套组合合成像在三个抽屉里各放几样积木:第一个抽屉是 m 种肽,第二个是 n 种连接子,第三个是 o 种羧酸。任取一样搭一样,能拼出的环肽数量是三者相乘(m × n × o)。真正省事的地方在于,搭完不必一个个拆开清洗,整盘直接拿去测——这才让成千上万成为可能。
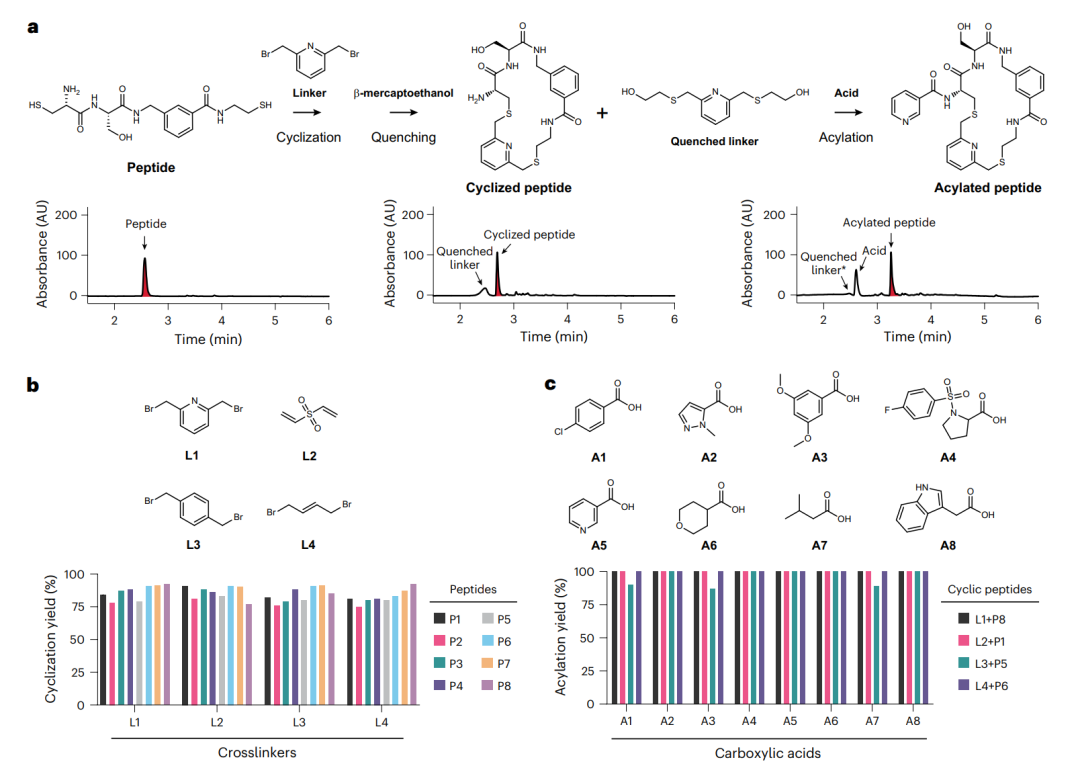
环化与酰化两步反应的效率验证。a 以一个示例肽展示完整流程:线性肽先环化、用 β-巯基乙醇淬灭多余连接子,再酰化,三张色谱图分别对应三步后的产物。b 是八个肽与四个连接子的环化产率,c 是酰化产率,可见两步反应对不同序列和试剂都有很宽的适应性。
环化与酰化两步反应的效率验证。a 以一个示例肽展示完整流程:线性肽先环化、用 β-巯基乙醇淬灭多余连接子,再酰化,三张色谱图分别对应三步后的产物。b 是八个肽与四个连接子的环化产率,c 是酰化产率,可见两步反应对不同序列和试剂都有很宽的适应性。
八千多个环肽,撞向凝血酶
有了合成方法,团队挑了凝血酶来练手。
〔名词小注:凝血酶(thrombin)〕一种胰蛋白酶样的丝氨酸蛋白酶,是血液凝固通路上的关键一环,也是抗血栓药物的重要靶点。已上市的口服凝血酶抑制剂达比加群酯就作用于它,但它在临床上的口服生物利用度只有 6.5%——它的活性形式带两个电荷,结合靶点离不开这两个电荷,可带电又过不了肠壁,只好把电荷用酯基包起来做成前药。
不带电的口服凝血酶抑制剂一直很难做。Heinis 团队以前做出来的凝血酶环肽,要么带电(穿不过膜),要么是二硫键环化的(不耐还原)。这次他们专门设计了一个库:不带任何电荷,用硫醚键环化。每个肽由六个构件组成——两个随机氨基酸、两个固定的巯基构件(如半胱氨酸)、一个随机连接子、一个随机外围羧酸。十个备选羧酸里包括 A11,它本身能微弱地结合凝血酶的 S1 口袋。
他们先合成了 384 个两端带巯基的短肽(四种格式理论上能组出 400 个,实际拿到 384 个),平均产量 4.4 微摩尔,不经纯化;再用两个连接子环化、十个羧酸(外加不加酸的对照)酰化,最终得到 8,448 个环肽,全部直接留在合成板里待筛(384 × 2 × 11 = 8,448)。这批环肽的物化性质分析显示,它们大多越出了五规则的范围——个头虽小,却和经典小分子明显不同。
筛选直接在合成板里做:往反应孔里加凝血酶和一种遇酶切割会发荧光的底物,把肽稀释到 10 微摩尔,测残余的凝血酶活性。8,448 个反应里,有 73 个(0.9%)把凝血酶活性压低了一半以上。把结果排成热图,会看到一个很整齐的规律:所有最活跃的肽,外围接的都是 A11 或 A12 这两个羧酸。对最强的 20 个命中肽重做一遍并复测,活性几乎不变,说明这套合成和筛选很稳。
对这 20 个肽做构效分析,归出六组结构相近的肽(11–30)。重新合成纯化后测抑制常数(Ki),都落在纳摩尔级,最好的是环肽 11,对人 α-凝血酶的 Ki 为 82 ± 10 纳摩尔。最强的第一组(11–16)共享一个 Cys-Phe 基序,半胱氨酸的氨基上接着氯噻吩。
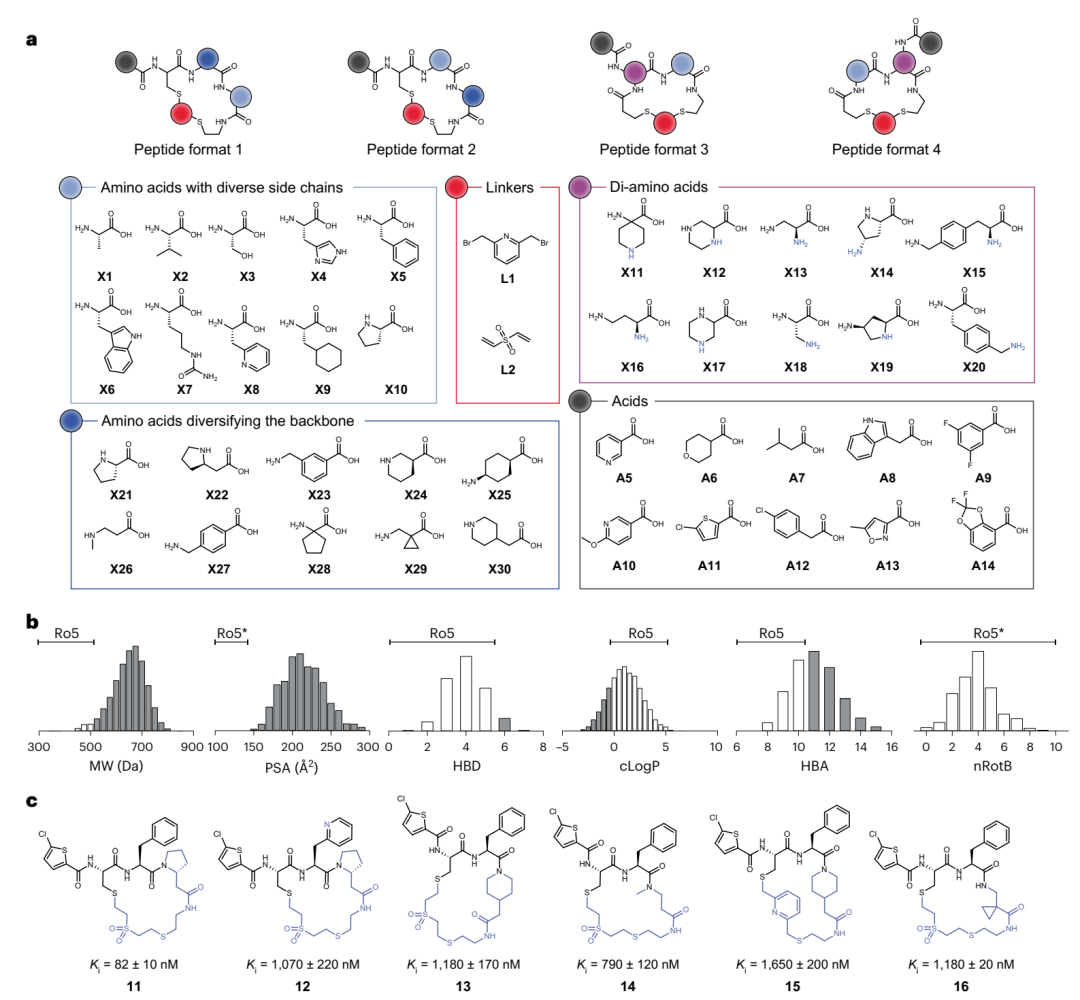
8,448 个环肽库的构成与凝血酶筛选。a 列出搭库用的全部积木:十个侧链各异的氨基酸、十个改变骨架的氨基酸、十个二氨基酸、两个连接子、十个羧酸,以及四种肽的格式。b 用直方图展示整个库的物化性质(分子量、极性表面积、氢键供体等),灰色部分是落在五规则之外的肽,可见绝大多数都出了框。c 是第一组六个最强命中肽(11–16)的结构和各自的 Ki,共同骨架用黑色标出。
8,448 个环肽库的构成与凝血酶筛选。a 列出搭库用的全部积木:十个侧链各异的氨基酸、十个改变骨架的氨基酸、十个二氨基酸、两个连接子、十个羧酸,以及四种肽的格式。b 用直方图展示整个库的物化性质(分子量、极性表面积、氢键供体等),灰色部分是落在五规则之外的肽,可见绝大多数都出了框。c 是第一组六个最强命中肽(11–16)的结构和各自的 Ki,共同骨架用黑色标出。
没有一个肽能三项全赢
找到强结合肽只是第一步。能不能口服,要看另外三项体外指标:扛不扛蛋白酶、能不能穿膜、在肝里被清除得快不快。团队把 20 个肽(11–30)一项项测了过去。
抗蛋白酶:把肽分别泡进模拟胃液(pH 1.2 的胃蛋白酶)和模拟肠液(pH 6.8 的胰酶),37 ℃ 放 8 小时。大多数肽都很稳——团队把这归因于它们多由非天然氨基酸组成、个头小、构象不易松动。
穿膜:用一种叫 PAMPA 的人工膜实验测被动扩散。
〔名词小注:PAMPA〕平行人工膜渗透实验。在两排孔之间隔一层涂了磷脂的人工膜,把化合物加在一侧(供体孔),看一段时间后有多少扩散到另一侧(受体孔),以此估计分子靠被动扩散穿过细胞膜的能力。
20 个肽的穿膜能力差异极大:有的很快扩散过去,有的几乎纹丝不动。换算成表观渗透系数(logPapp),从完全不透的约 −8 到完全透的约 −5 都有,最好的六个在 −6.0 到 −5.2 之间,已经接近做参照的华法林(−5.3)。
代谢稳定:还是用大鼠肝微粒体。结果只有五个肽比维拉帕米清除得更慢——团队把维拉帕米这个清除偏快的口服药,定为能接受的稳定性下限。
把三项放在一起看,结论很清楚:每一项里都有几个肽表现不错,却没有任何一个肽能在三项上全部过关。
〔难在哪〕这就是口服肽开发的核心矛盾被摊开的一刻。强结合、能穿膜、耐代谢,像一个三角形的三个角,顾此往往失彼;一个分子在某一项上做到极致,常常是在另一项上让了步。接下来的全部工作,本质上都是在这个三角形里来回腾挪。
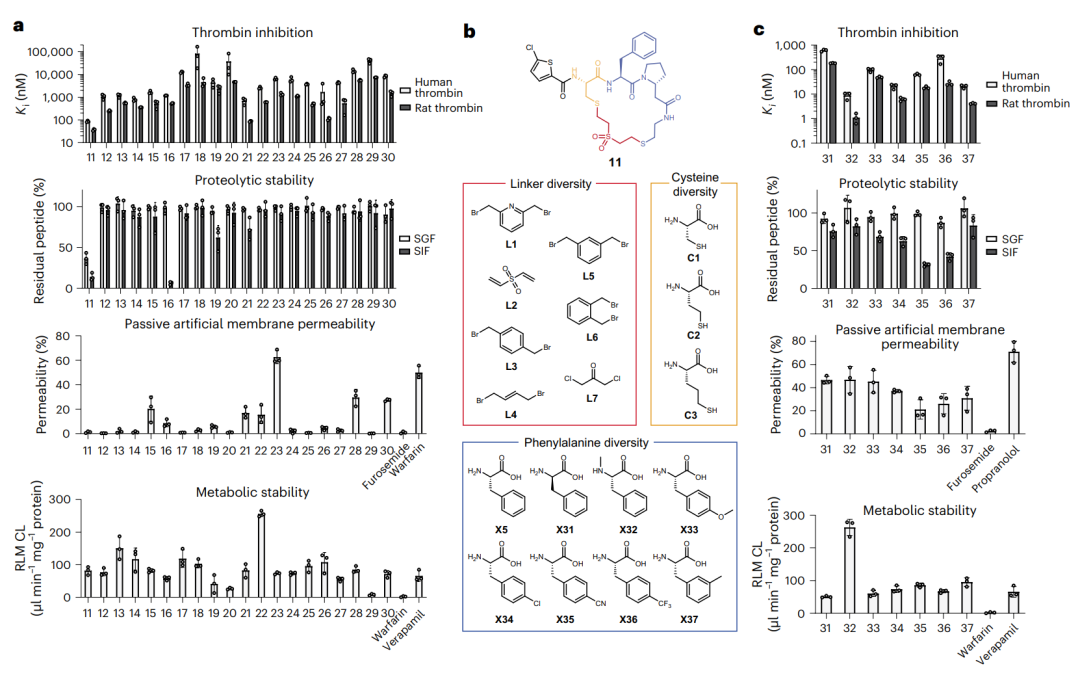
凝血酶抑制肽的体外表征与子库筛选。a 把肽 11–30 的四项指标(对人和大鼠凝血酶的抑制、抗胃肠液降解、人工膜渗透、肝微粒体代谢稳定)逐一排开,可一眼看出没有哪个肽四项都好。b 是基于环肽 11 搭建子库时,在苯丙氨酸、连接子、半胱氨酸三个位置上替换用的积木。c 是优化后一批肽(31–37)的同样四项表征。
凝血酶抑制肽的体外表征与子库筛选。a 把肽 11–30 的四项指标(对人和大鼠凝血酶的抑制、抗胃肠液降解、人工膜渗透、肝微粒体代谢稳定)逐一排开,可一眼看出没有哪个肽四项都好。b 是基于环肽 11 搭建子库时,在苯丙氨酸、连接子、半胱氨酸三个位置上替换用的积木。c 是优化后一批肽(31–37)的同样四项表征。
一轮一轮,把短板补上
既然没有现成的赢家,就从最强的肽 11 出发,一轮轮改。高效的合成让这种小改快测几乎没有成本。
第一轮,团队保留肽 11 上的 β-高脯氨酸和氯噻吩不动,只在另外三个位置(半胱氨酸、苯丙氨酸、连接子)上换不同的积木,搭了一个 168 个成员的子库(8 种苯丙氨酸 × 7 种连接子 × 3 种半胱氨酸)。这一轮还用上了一个聪明的筛选设计:把粗反应直接加进 PAMPA 板的供体孔,然后同时测供体孔和受体孔里的凝血酶抑制活性。
〔Aha 时刻〕一次实验,两个答案。供体孔里测到的抑制活性,反映这个肽还结不结合凝血酶;受体孔里测到的抑制活性,反映它有没有穿过那层膜扩散过去。同一块板,同时读出还有没有活性和穿没穿得过去这两项最关键的性质——把原本要分开做的两件事叠在了一起。
读数告诉他们:换掉苯丙氨酸会大幅削弱活性,但换半胱氨酸侧链和连接子都还能接受;受体孔显示,用 L1 和 L4 连接子环化的肽,比原来用砜基连接子 L2 的穿膜性好得多,其中 L4 在活性和穿膜上都最好。挑出来纯化的肽 31–37 里,最强的是肽 32:对人和大鼠凝血酶的 Ki 分别是 8.9 和 1.10 纳摩尔,比母体肽 11 强了十倍。它抗蛋白酶、能穿膜,几乎都达标了,只有一项例外:肽 32 在肝微粒体里被清除得异常快(260 微升/分钟/毫克),代谢稳定性太差,没法进体内实验。
问题又回到了硫醚键。对肽 32 做质谱分析,看到随孵育时间增加的 +16 和 +32 质量峰,指向硫醚键上的氧化,很可能形成了亚砜。既然硫原子是软肋,那就把它整个拿掉。第二轮,团队用全碳氢的连接子重新把肽 32 环化,靠的是固相上的关环复分解(RCM)。
〔名词小注:关环复分解(RCM)〕一种用金属催化剂把两段末端带碳碳双键的链连成一个环的反应。在这里,它让环肽的环不再靠含硫的硫醚键闭合,而改用纯碳碳骨架——彻底躲开了硫原子被氧化的问题。
七个用 RCM 环化的肽 38–44 里,最好的是肽 43:对人和大鼠凝血酶的 Ki 为 50 和 11.1 纳摩尔,代谢稳定性比肽 32 提高了三倍(清除率 85),而且依然能穿膜。到这一步,肽 43 终于够格做体内实验了。
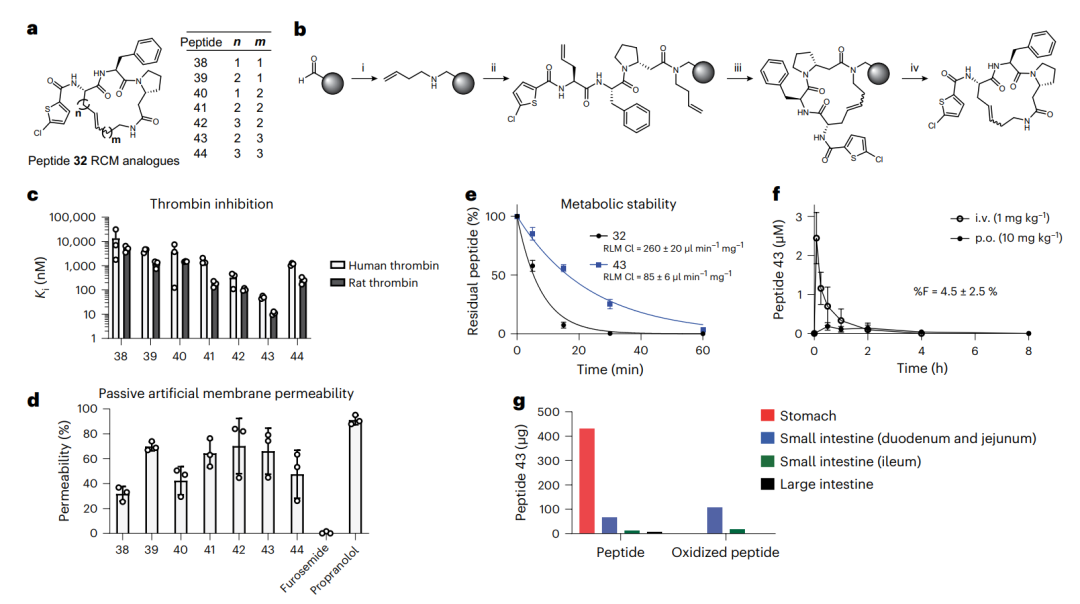
用碳氢连接子环化的肽(基于肽 32)。a 是七个用 RCM 环化的肽 38–44 的结构,连接子长度各不相同。b 画出 RCM 在固相上的合成步骤。e 把肽 32 与它的 RCM 类似物 43 在肝微粒体里的稳定性放在一起对比,43 明显更稳。f 是肽 43 在大鼠里的口服与静脉血药曲线,g 显示口服后它在胃肠各段里大部分仍完整,但已能检出少量被氧化的肽。
用碳氢连接子环化的肽(基于肽 32)。a 是七个用 RCM 环化的肽 38–44 的结构,连接子长度各不相同。b 画出 RCM 在固相上的合成步骤。e 把肽 32 与它的 RCM 类似物 43 在肝微粒体里的稳定性放在一起对比,43 明显更稳。f 是肽 43 在大鼠里的口服与静脉血药曲线,g 显示口服后它在胃肠各段里大部分仍完整,但已能检出少量被氧化的肽。
灌进大鼠的胃里
把肽 43 灌进大鼠的胃里(n=3),血样显示它确实进了血液循环:30 分钟后血浆峰浓度 183 纳摩尔,口服生物利用度 4.5% 。作为阳性对照,他们用了 Lokey 实验室设计的、专门做得能穿膜的环六肽 1NMe3,它在同样条件下的口服生物利用度是 27% 。
4.5% 还不够。为了弄清肽 43 在体内到底经历了什么,团队又给大鼠灌了一次药,30 分钟后取出胃、小肠(十二指肠和空肠、回肠分开)、大肠分别分析。大部分肽是完整的,但在消化道里就已经能找到一些被氧化的肽(多出 16、32 的质量)。这说明只要把残余的氧化位点再堵上,生物利用度还能往上走。
第三轮,目标是干掉肽 43 上最后那个氧化位点。质谱没能精确定位氧化在哪(找不到那个被氧化的碎片),但团队怀疑是氯噻吩这个基团。于是他们又做了一轮快速的替换:固定环的部分,只在外围氨基上换不同的羧酸。考虑到肽 43 很可能结合在凝血酶的 S1 口袋里,他们挑了 17 个同样能塞进 S1 口袋的羧酸来试,氯噻吩本身作阳性对照。
筛出来的肽 45 和 46 活性都和母体 43 相当。略强的肽 46 用 4-氯苯甲酸替掉了氯噻吩,对人和大鼠凝血酶的 Ki 为 65 和 10.3 纳摩尔,抗蛋白酶、能穿膜都很好,在肝微粒体里又比 43 稳了约一倍。专一性测试表明,肽 46 只选择性地抑制凝血酶(对其他几种胰蛋白酶样蛋白酶基本不碰),而且是竞争性抑制剂。最后测口服:肽 46 的血浆峰浓度达到 400 纳摩尔,是肽 43 的两倍;口服生物利用度升到 18.4% ,已逼近那个专门设计来穿膜的参照肽 1NMe3 的 27%。
从肽 11 到肽 32(强十倍),到肽 43(换掉硫醚、能进体内),再到肽 46(堵上最后的氧化位点、口服 18%),这条优化路径上的每一轮,都精确地针对上一轮暴露出的那个短板。
〔一点判断〕18.4% 这个数字,放在肽类药物里相当亮眼。肽 46 是从零设计、完全不带电、针对一个具体靶点造出来的小环肽,在大鼠里的口服生物利用度比同样在大鼠中测试的达比加群酯高出约 4.5 倍(达比加群酯之所以能口服,是靠把两个电荷用酯基包成前药)。它还没到可以做药的程度,但已经证明:从头造一个能口服的靶向环肽,这条路走得通。
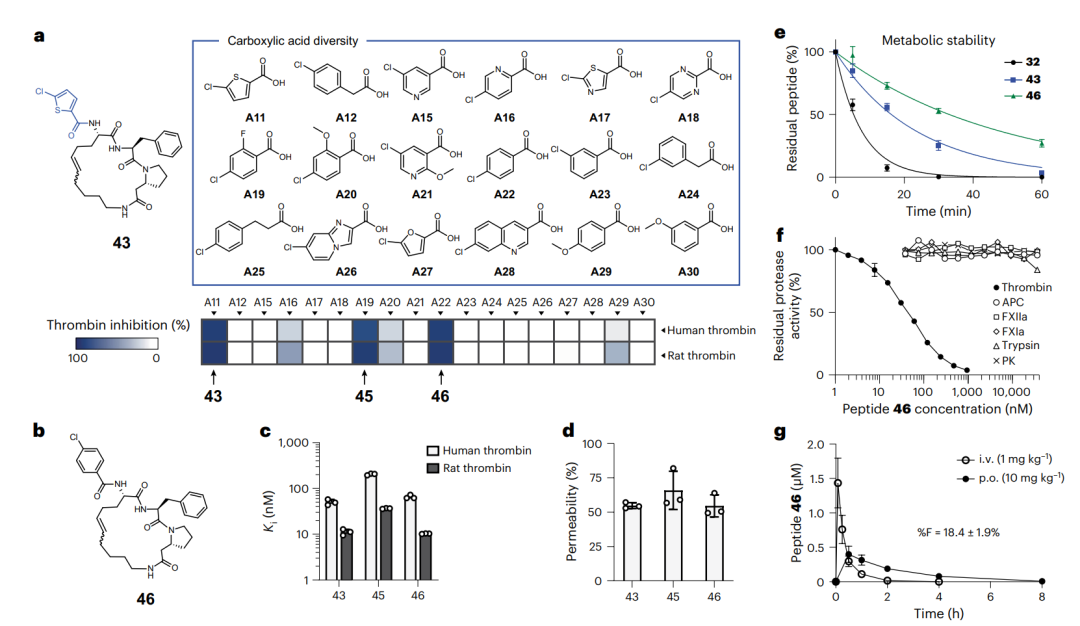
代谢更稳、口服更好的环肽 46。a 是基于肽 43、把外围氯噻吩换成 17 个类似羧酸的子库,下方热图是它们对人和大鼠凝血酶的抑制强度。b 是肽 46 的化学结构。e 把肽 32、43、46 三者在肝微粒体里的稳定性放在一起比较,可见逐代改善。f 是肽 46 的专一性谱,只有凝血酶被显著抑制。g 是肽 46 在大鼠里的口服与静脉血药曲线,口服生物利用度 18.4%。
代谢更稳、口服更好的环肽 46。a 是基于肽 43、把外围氯噻吩换成 17 个类似羧酸的子库,下方热图是它们对人和大鼠凝血酶的抑制强度。b 是肽 46 的化学结构。e 把肽 32、43、46 三者在肝微粒体里的稳定性放在一起比较,可见逐代改善。f 是肽 46 的专一性谱,只有凝血酶被显著抑制。g 是肽 46 在大鼠里的口服与静脉血药曲线,口服生物利用度 18.4%。
这套方法能做什么,不能做什么
这项工作真正的产出,不只是一个口服 18% 的凝血酶抑制肽,而是一整套能反复使用的造肽方法:用硫醚环化拿到能不纯化直接筛的大库,用声波分液把成千上万个环肽组合出来,再用能同时读出结合与穿膜的子库筛选,一轮轮把候选肽往口服的方向推。这套方法对任何有功能读数的靶点都适用。
但作者把局限讲得很清楚。
〔边界〕第一,硫醚键到底能不能直接做药,作者明说自己也拿不准。一方面,好几个已上市的口服药本身就含硫醚键,他们的一些测试肽稳定性也超过了已上市药;另一方面,他们最强的几个肽偏偏卡在硫醚的氧化上,不得不换掉。结论是稳定性很可能取决于具体序列,得一例一例看。一个更彻底的办法是从一开始就用 RCM 的全碳环,省掉后面换连接子这步麻烦事——但要把 RCM 做到能高效环化大量不同的肽,还需要不少开发工作。
第二,这个肽还不能当药。要达到药理活性所需的亚纳摩尔级强度还差一截;作者用的全是市售构件,认为换上定制构件应能进一步提升。另外,RCM 环上那个碳碳双键的几何构型并没有定准,肽 46 本身就是一对 E/Z 异构体的混合物。
第三,目前的全部体内证据都来自大鼠,靶点也只有凝血酶一个。方法的通用性是论证出来、加上一个成功案例支撑的,还没有在更多靶点、尤其是更难的靶点(比如蛋白-蛋白相互作用)上得到检验。
不带电这一点,作者特别看重。大多数凝血酶抑制剂都要靠一个带电基团去结合 S1 口袋,而带电几乎注定难口服;这批肽干脆不带电,从源头上让开了麻烦——这也是它能比达比加群酯口服更好的根本原因。
把针剂变成药片
很长一段时间里,肽类药物被困在一个尴尬的位置:它们结合靶点的本领一流,却几乎都得用针头送进身体。这篇工作没有声称解决了所有问题——它造出的肽离真正的药还有距离,证据也还停在大鼠和单一靶点上。但它把一件原本要靠运气和巨量手工劳动才能偶得的事,变成了一个可以一轮轮逼近目标的过程:每暴露一个短板,就有办法去补;每补一次,就离口服更近一步。
如果这套方法的简单和稳健真像作者期待的那样被广泛用起来,那些至今只能靠打针、甚至根本无药可用的难缠靶点,也许有一天会等来一颗能直接吞下去的小环肽。把针剂变成药片,从来不只是方便那么简单——它决定了一种药能不能走进那些本来够不着它的人的日常。
参考文献
原文 De novo development of small cyclic peptides that are orally bioavailable(Merz、Habeshian、Li 等,通讯作者 Christian Heinis,EPFL),发表于 Nature Chemical Biology 2024 年第 20 卷 624–633 页,DOI 10.1038/s41589-023-01496-y。
本文为开放获取(CC BY 4.0),原始数据随论文一并提供;利益声明中,通讯作者等为衍生公司 Orbis Medicines 的创始人。

本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号