BioMatrix: 一个模型、一套 token,真正原生统一生物序列、结构与语言

BioMatrix: 一个模型、一套 token,真正原生统一生物序列、结构与语言

DrugOne
发布于 2026-07-06 15:37:44
发布于 2026-07-06 15:37:44

生物大模型,为什么还缺一块“拼图”?
近年来,基础大模型正在加速进入生命科学与药物研发领域。从分子生成、蛋白质设计,到分子-蛋白相互作用预测,AI 正在帮助研究者更快地理解复杂生命系统。然而,生物世界并不是由单一模态构成的。
一个分子,既可以被表示为 SMILES 或 SELFIES 这样的序列,也可以被描述为三维构象,还可以出现在论文、专利或数据库的自然语言描述中;一个蛋白质,既有氨基酸序列,也有空间结构,还关联着功能、疾病、通路与实验背景。更重要的是,真实的生物任务往往不是“单个实体”的问题,而是分子与蛋白、蛋白与蛋白之间的相互作用问题。
现有生物基础模型通常只能覆盖其中的一部分:有的模型擅长序列与文本建模,却缺少显式结构能力;有的模型能够处理结构,却局限于蛋白或单一实体;还有一些系统依赖外部编码器、适配器或专门输出头,导致“能读某种模态,却不能原生生成这种模态”。
换句话说,现有生物大模型像一支各司其职的专家团队——有人懂序列,有人懂结构,有人懂文本,但它们之间需要翻译官(适配器/编码器)才能协作。BioMatrix 想做的是:让一个人同时掌握所有能力,不需要翻译。
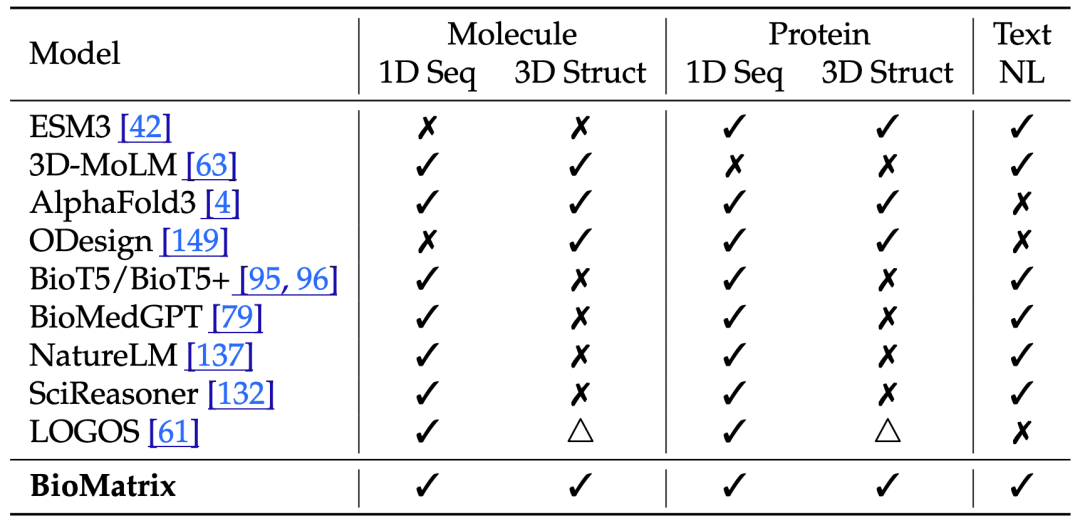
为了补上这块关键拼图,我们提出 BioMatrix [1]:一个面向分子与蛋白的统一多模态生物基础模型。BioMatrix 将分子序列、分子结构、蛋白序列、蛋白结构与自然语言统一到同一个离散 token 空间中,在单一 decoder-only 架构下完成理解与生成,推动生物大模型从“多组件拼接”走向“原生统一建模”。
BioMatrix 统一多模态 Tokenization
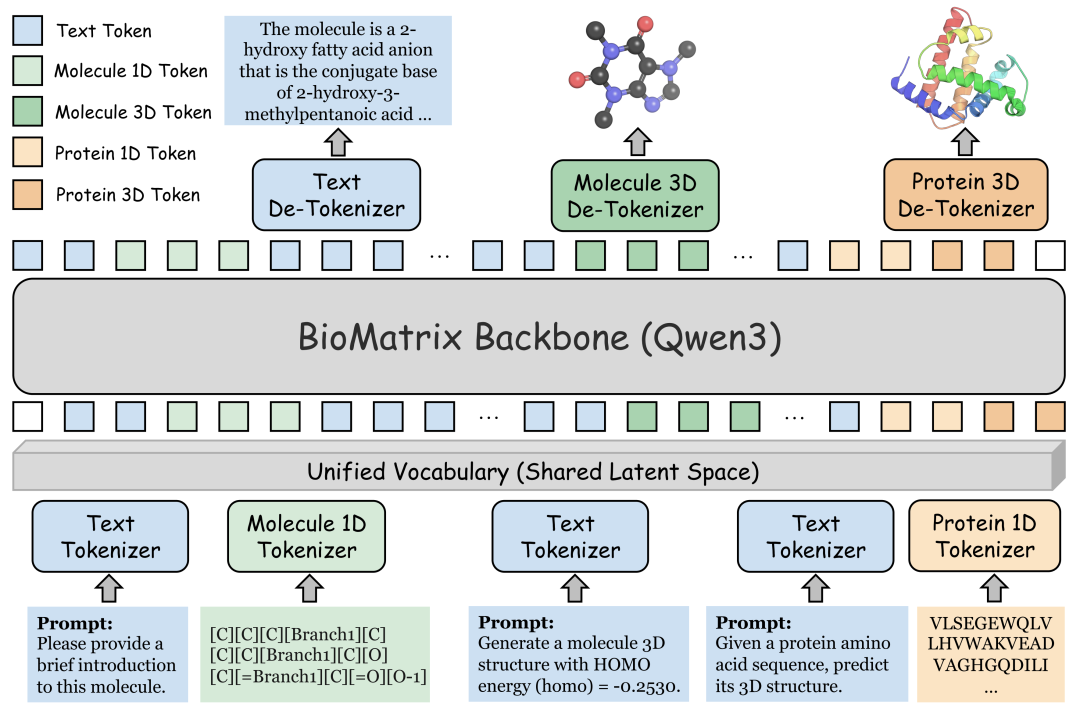
BioMatrix 的核心思想可以概括为一句话:把分子、蛋白与文本放进同一个“模态矩阵”中,让模型用同一种语言理解和生成生命科学对象。
具体而言,BioMatrix通过统一多模态 tokenization,将以下五类信息映射到共享离散词表中:
- 分子1D序列:支持 SMILES 与 SELFIES 表示;
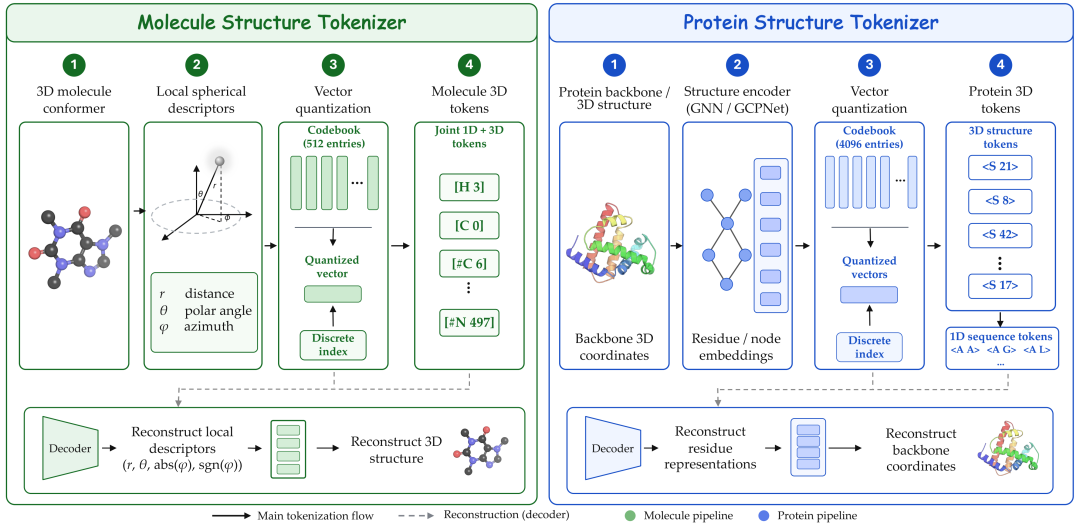
- 分子3D结构:通过改进的 MolStrucTok [2] 将三维构象离散化;
- 蛋白1D序列:使用氨基酸序列 token 表示;
- 蛋白3D结构:通过 GCP-VQVAE [3] 将蛋白结构编码为离散结构 token;
- 自然语言文本:保留语言模型原有文本词表,用于承载科学描述、任务指令与知识背景。
在这个框架下,过去看似完全不同的任务——例如分子描述生成、文本条件分子设计、蛋白 folding、inverse folding、蛋白序列-结构联合生成、分子-蛋白亲和力预测——都可以被统一为同一种形式:给定一段由不同模态 token 组成的输入,让模型继续生成目标模态 token。
这意味着 BioMatrix 不需要额外的外部编码器、投影适配器或任务专用输出头。它像通用语言模型生成文本一样,原生处理分子、蛋白、结构与语言之间的转换和推理。
从“单模态专家”到“多模态通才”
过去的生物 AI 模型往往像不同领域的专家:有的专攻蛋白结构,有的专攻分子生成,有的专攻生物医学文本理解。但真实科研问题更像一个跨学科项目,需要同时读懂分子结构、蛋白结构、实验文本和相互作用关系。
BioMatrix 试图把这些能力装进一个统一模型中。它的目标不是在某一个任务上定制一个专用系统,而是构建一个可迁移、可扩展的生物基础模型底座,让同一个模型能够覆盖更广泛的生命科学任务。
相比 BioT5 系列 [4,5] 采用的多模块架构,BioMatrix 迈向了原生统一:所有模态共享同一套 token 空间和单一 decoder,不再需要外部编码器或适配器来桥接不同模态。
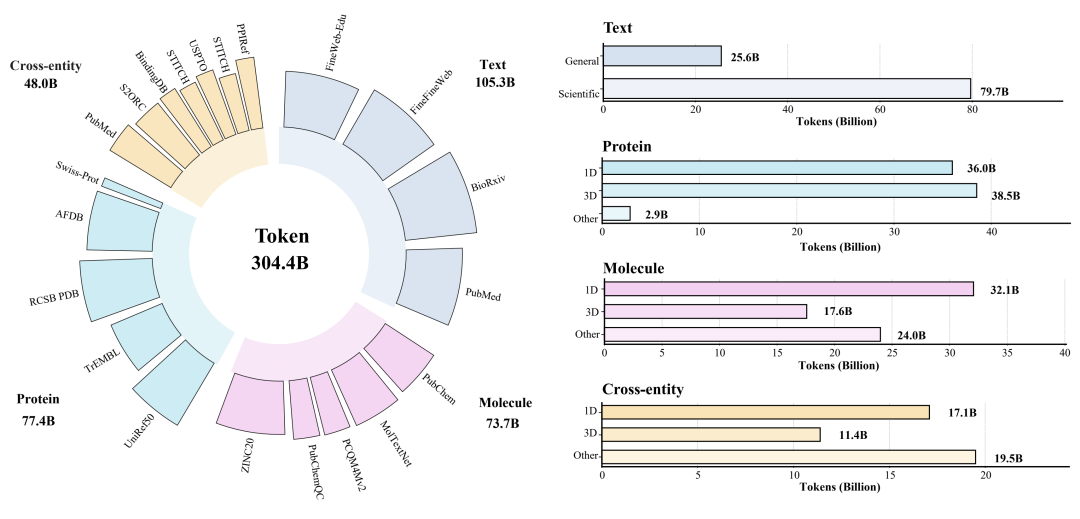
为此,BioMatrix 基于 Qwen3-1.7B 和 Qwen3-4B [6] 构建了两个规模的模型,并在约 304.4B tokens 的大规模多模态语料上进行继续预训练。该语料覆盖四类核心数据:
- 通用与科学文本;
- 分子相关数据,包括分子序列、结构与文本视图;
- 蛋白相关数据,包括蛋白序列、结构与功能描述;
- 跨实体与交错数据,包括分子-蛋白相互作用、蛋白-蛋白相互作用,以及生物实体与科学文本交织的数据。
通过这样的训练,模型不仅学习单个实体的表示,也学习不同模态、不同实体之间的对应关系。
指令微调:覆盖80个生物下游任务
为了验证 BioMatrix 是否真的具备统一建模能力,我们构建了一个覆盖面广泛的指令微调与评测体系,涵盖 80 个下游任务、6 大任务类别。这些任务横跨三类实体范围:
- 分子任务:包括分子描述生成、文本条件分子设计、分子性质预测、分子构象生成等;
- 蛋白任务:包括蛋白 folding、inverse folding、蛋白功能注释、蛋白序列-结构联合生成等;
- 分子-蛋白 / 蛋白-蛋白交互任务:包括分子-蛋白亲和力预测、蛋白-蛋白结合界面预测、跨实体相互作用建模等。
同时,每类任务又进一步覆盖 1D 序列与 3D 结构两种模态层级。
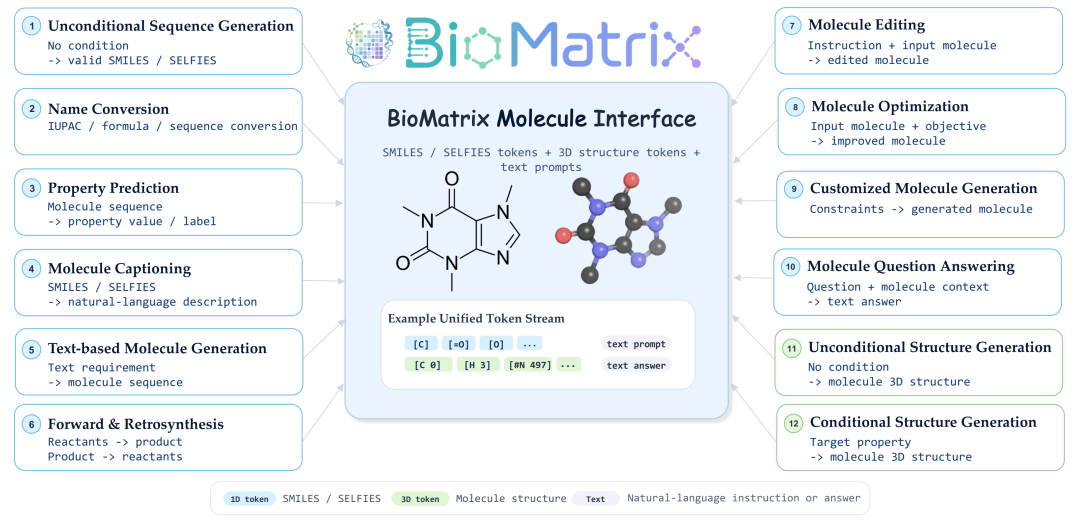
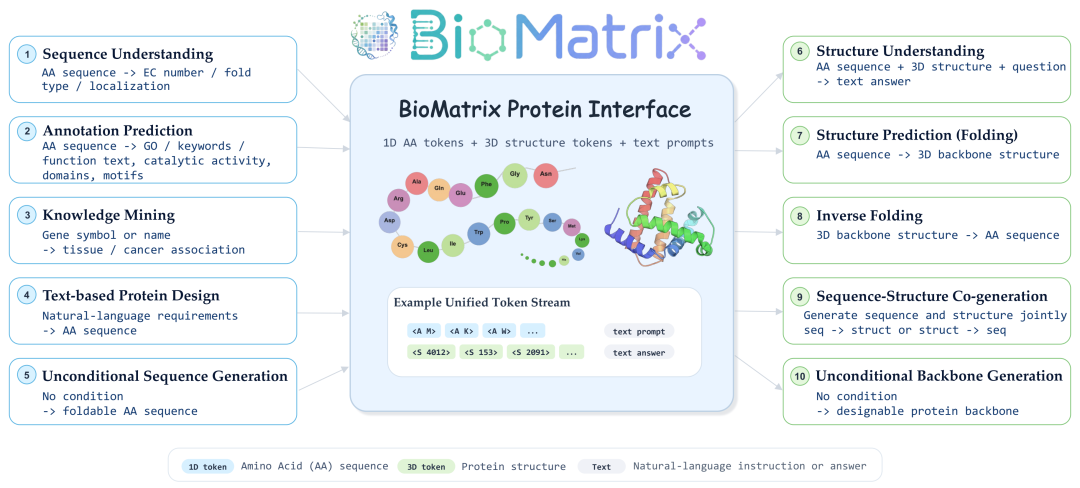
这套任务体系不仅考察模型是否“会做题”,更考察它能否在序列、结构、文本和跨实体关系之间自由切换。
结果突出,一个统一模型,在77/80个任务上达到领先或竞争水平
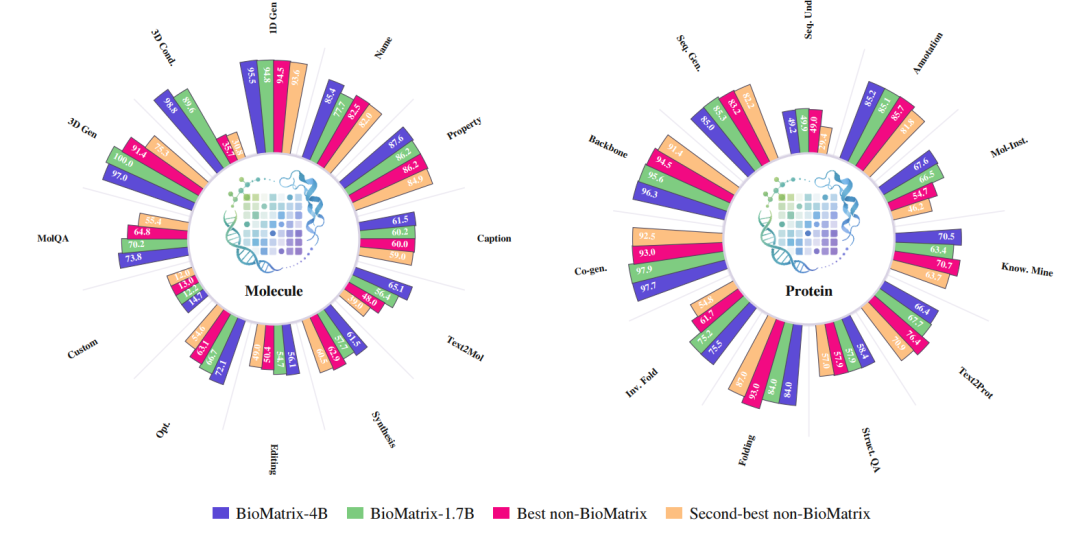
实验结果表明,BioMatrix 在广泛的生物任务上展现出强大的通用能力。在 80 个评测任务中,BioMatrix 在 77 个任务 上达到 SOTA 或具备竞争力的表现,证明单一原生多模态通才模型可以在大量任务上匹配甚至超越专用模型。
更值得关注的是,BioMatrix 的优势尤其体现在那些真正需要“统一建模”的任务上,例如:
- 文本条件分子生成:模型需要把自然语言中的设计意图转化为分子表示;
- 属性条件分子构象生成:模型需要同时理解分子结构与目标性质;
- 蛋白序列-结构联合生成:模型需要在序列与三维结构之间建立一致关系;
- 结构驱动的分子-蛋白亲和力预测:模型需要同时处理小分子结构、蛋白结构与跨实体相互作用。
这些结果说明,BioMatrix 的提升并不仅来自更大的模型规模,而来自其统一 token 空间和原生多模态建模方式。
结论与未来展望
BioMatrix 提出了一条通向统一生物基础模型的新路径:不是为每个任务构造一个专用架构,而是将分子、蛋白、结构和语言纳入同一个 token 空间,让模型在统一生成式框架下学习生命科学对象之间的关系。例如,在药物发现中,研究者可以用自然语言描述目标性质,让模型直接生成候选分子及其 3D 构象,同时预测与靶点蛋白的亲和力——全在一个模型内完成,无需串联多个工具。
当然,BioMatrix 仍然有进一步提升空间。例如,当前分子 3D 结构与蛋白 3D 结构仍使用不同结构 tokenizer,尚不能完全原生表达小分子与蛋白口袋之间的相对空间位姿;模型目前主要覆盖小分子与蛋白,还没有扩展到 DNA、RNA、糖类、脂质等更多生物实体;同时,如何在高度异构且不平衡的生物任务中训练一个真正全任务统一的 SFT 模型,也仍是值得探索的问题。
尽管如此,BioMatrix 已经迈出了关键一步:它证明了一个统一的、原生多模态的生物大模型,可以在广泛任务上接近或超越专用模型,为 AI4Science、药物发现和蛋白工程打开了新的想象空间。
- 投稿人:裴启智、吴郦军
- 投稿单位:上海人工智能实验室、中国人民大学
- 论文:https://arxiv.org/abs/2606.22138
- 项目:https://github.com/QizhiPei/biomatrix
- 模型与数据: https://huggingface.co/collections/QizhiPei/biomatrix
参考资料
[1] Pei, Qizhi, et al. "BioMatrix: Towards a Comprehensive Biological Foundation Model Spanning the Modality Matrix of Sequences, Structures, and Language." arXiv preprint arXiv:2606.22138 (2026).
[2] Gao, Kaiyuan, et al. "Tokenizing 3d molecule structure with quantized spherical coordinates." Proceedings of the 32nd ACM SIGKDD Conference on Knowledge Discovery and Data Mining V. 1. 2026.
[3] Pourmirzaei, Mahdi, et al. "GCP-VQVAE: A Geometry-Complete Language for Protein 3D Structure." NeurIPS 2025 AI for Science Workshop.
[4] Pei, Qizhi, et al. "Biot5: Enriching cross-modal integration in biology with chemical knowledge and natural language associations." Proceedings of the 2023 Conference on Empirical Methods in Natural Language Processing. 2023.
[5] Pei, Qizhi, et al. "Biot5+: Towards generalized biological understanding with iupac integration and multi-task tuning." Findings of the Association for Computational Linguistics: ACL 2024. 2024.
[6] Yang, An, et al. "Qwen3 technical report." arXiv preprint arXiv:2505.09388 (2025).
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号