分析梳理--linux实现虚拟筛选
原创
分析梳理--linux实现虚拟筛选
原创
追风少年i
发布于 2026-04-13 12:49:00
发布于 2026-04-13 12:49:00
作者,Evil Genius
今天我们来实现虚拟筛选。
第一步,处理蛋白质PDB文件,注意这时候的PDB分子已经进行了去水处理(pymol),其中去除水分子的注意事项。
当PDB文件中含有水分子时,必须将其删除,否则会严重影响对接结果的准确性。
水分子的存在会带来两个核心问题:
物理位阻:水分子会占据结合口袋的空间,导致配体分子无法正确进入。
能量计算错误:对接软件会将水分子视为蛋白的一部分,产生虚假的氢键和排斥力。
正确处理流程
正确做法是:在生成PDBQT文件前,删除所有水分子(通常以 HOH 或 WAT 标识),但需保留可能对催化或结合至关重要的结构性水分子(数量极少,通常≤3个)。
推荐流程:
使用PyMOL或Chimera等可视化软件查看结构,定位结合口袋。
删除所有非关键水分子。
保留关键水分子:判断依据可查看同源蛋白结构文献,或通过PyMOL观察该水分子是否与蛋白/配体形成稳定氢键网络。
保存为新的PDB文件,再用于生成PDBQT。
去除水分子的前后对比
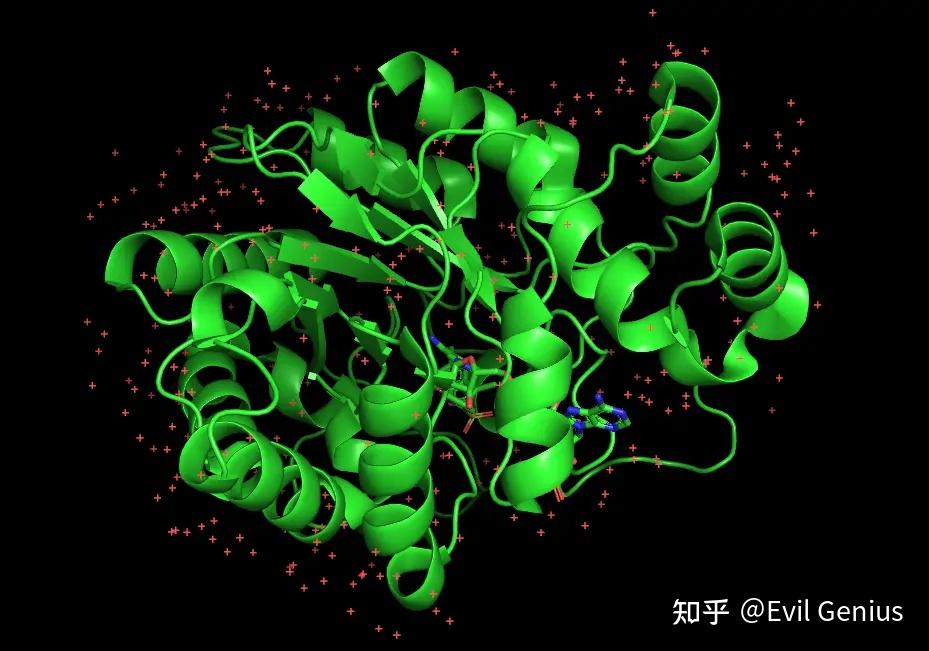
去水前
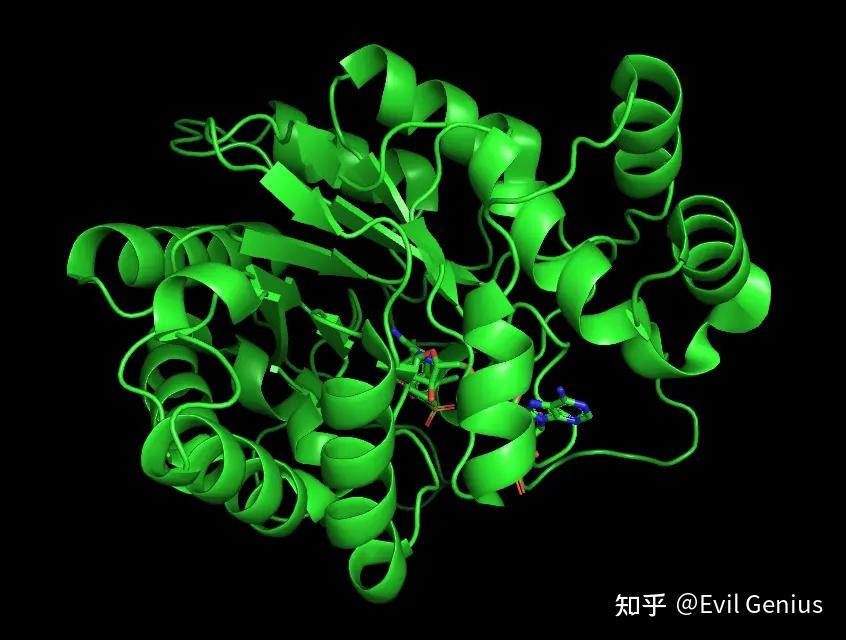
去水后
第一步:生成的蛋白质PDB文件,加氢,加电荷,生成PDBQT
prepare_receptor.py -r protein_no_water.pdb -o protein.pdbqt -A hydrogens但是我们最好联合reduce使用,因为
特性 | prepare_receptor.py -A hydrogens | reduce |
|---|---|---|
加氢范围 | 添加所有氢原子(调用PyBabel) | 添加所有氢原子,并优化氢键网络 |
侧链优化 | 无 | 自动翻转Asn/Gln/His侧链,形成最优氢键网络 |
输出格式 | 直接输出PDBQT(含原子类型和电荷) | 输出PDB格式,还需再转PDBQT |
适用场景 | 常规分子对接(一键完成) | 高精度对接、分子动力学模拟前处理 |
所以完整的代码是
reduce -FLIP protein_no_water.pdb > protein_h.pdb
# 转换为 PDBQT 格式(必须使用 pythonsh)
prepare_receptor -r protein_h.pdb -o protein.pdbqt -A hydrogens这个过程实现了
对蛋白质分子的合并非极性氢(AutoDock/Vina 力场要求)
为所有原子分配正确的 AutoDock 原子类型(如 A、NA、OA 等)
计算并写入 Gasteiger 部分电荷
第二步:对配体分子的处理
prepare_ligand.py -l ligand.pdb -o ligand.pdbqt -A hydrogens这个输入的格式包括pdb、mol2、sdf 等格式。
批量处理也很简单
for ligand in *.pdb; do
name=$(basename "$ligand" .pdb)
prepare_ligand.py -l "$ligand" -o "${name}.pdbqt" -A hydrogens
done第三步:定义柔性残基(还是以41位的组氨酸和145的半胱氨酸为例)
用法
prepare_flexreceptor.py -r receptor_filename -s list_of_names_of_residues_to_move例子
prepare_flexreceptor.py -r protein.pdbqt -s HIS41_CYS145完成柔性残基的定义后,将分别生成rigid区域和flex区域的pdbqt文件:
proH_rigid.pdbqt 刚性部分
proH_flex.pdbqt 柔性侧链部分
第四步:设置对接盒子
示例如下,contig.txt
receptor = receptor.pdbqt
center_x = 15.0
center_y = 20.0
center_z = 10.0
size_x = 20.0
size_y = 20.0
size_z = 20.0
exhaustiveness = 32
num_modes = 9
energy_range = 3.0其中参数的意义
exhaustiveness = 32 (搜索 exhaustive-ness,即搜索彻底性)
直译:穷举性、彻底性。
作用:这是最重要的参数,控制着对接搜索的强度或努力程度。
具体含义:值越大,程序在配体-受体结合口袋中进行的全局搜索次数就越多。这能增加找到真正全局最优结合构象的概率,但同时会显著增加计算时间(几乎与设定值成正比)。
取值范围与建议:
默认值:8。
快速筛选/测试:1 - 4(速度极快,用于排除明显不结合的分子,但可能漏掉最优解)。
常规对接:8 - 32(在精度和速度之间取得良好平衡。8是官方默认值,32已经是相当高的精度)。
高精度/最终确认:32 - 64或更高(用于已筛选出的几个关键分子,追求最可靠的结果)。
核心原则:exhaustiveness 增加一倍,运行时间也大约增加一倍。建议先用小值(如4或8)快速测试流程,正式运行时再提高。
energy_range = 3.0 (Energy Range,即能量范围)
直译:能量范围。
作用:与 num_modes 配合使用,用于控制输出构象之间的能量差异。
具体含义:程序只会输出那些结合能(Affinity)与最优构象(排名第一)的差值 ≤ energy_range 的构象。超过这个能量差的构象,即使排名再靠前,也不会被输出。
取值范围与建议:
默认值:3.0 (单位:kcal/mol)。
原理:这是一个过滤机制。例如,如果最优结合能为 -9.0 kcal/mol,设置 energy_range = 3.0,那么 Vina 只会输出结合能优于 -6.0 kcal/mol 的构象。即使你设置了 num_modes = 9,但排名第9的结合能是 -5.8 kcal/mol(差值3.2 > 3.0),它也不会被输出,实际输出可能少于9个。
建议:
默认 3.0 即可,足以覆盖大多数有意义的构象变化。
如果只想看能量最低的几个极近似构象,可以减小到 1.0 或 1.5。
如果想看到更多样化(但能量可能更高)的结合模式,可以增大到 4.0 或 5.0。
第五步:执行分子对接
vina --config config.txt --receptor proH_rigid.pdbqt --flex proH_flex.pdbqt --ligand ligand.pdbqt --out out.pdbqt批量对接呢?
#!/bin/bash
# 刚性受体和柔性残基文件
RECEPTOR_RIGID="proH_rigid.pdbqt"
RECEPTOR_FLEX="proH_flex.pdbqt"
CONFIG_FILE="config.txt"
# 配体文件夹和输出目录
LIGAND_DIR="./ligands"
OUTPUT_BASE_DIR="./results"
# 创建输出目录
mkdir -p $OUTPUT_BASE_DIR
# 批量对接
for ligand_file in $LIGAND_DIR/*.pdbqt; do
ligand_name=$(basename "$ligand_file" .pdbqt)
output_dir="$OUTPUT_BASE_DIR/$ligand_name"
mkdir -p $output_dir
echo "正在对接: $ligand_name"
vina --config $CONFIG_FILE \
--receptor $RECEPTOR_RIGID \
--flex $RECEPTOR_FLEX \
--ligand $ligand_file \
--out "$output_dir/out.pdbqt" \
--log "$output_dir/log.txt"
done
echo "批量对接完成!"其中使用autodock和vina力场的区别。
对比维度 | AutoDock (特指AutoDock 4) | AutoDock Vina |
|---|---|---|
模型类型 | 基于物理的力场(Physics-based Force Field) | 经验性 + 知识基础(Empirical + Knowledge-based) |
能量项构成 | - 范德华力 (Lennard-Jones 12-6)- 氢键 (方向性 12-10 势)- 静电作用 (库仑势)- 去溶剂化能 (基于电荷的成对项)- 构象熵罚 | - 范德华类势 (引力高斯函数 + 排斥项)- 非定向氢键项- 疏水项- 构象熵罚 |
特点 | 物理模型更完整,考虑了静电和溶剂效应,因此计算量更大、速度较慢 。 | 针对速度和准确性进行了高度优化,删去了静电和显式去溶剂化项,用更简单的经验项替代,使得速度比AutoDock快数十倍到上百倍 |
深入解读:两种力场的差异
虽然它们都能用来预测分子间结合模式,但背后的“方法论”完全不同:
- AutoDock 4 系列:追求“还原”的物理模型 AutoDock 4 的力场更像是一个精简版的分子力学力场,试图直接计算物理相互作用。它包含了静电能和去溶剂化能,使得对结合自由能的预测在理论上更“完整”。这种方法的优点是准确性高,但代价是计算复杂,速度较慢。为了应对特殊场景,它还发展出了专门针对含锌金属蛋白的 AutoDock4Zn 力场,以及针对多环化合物的处理方案。
- AutoDock Vina:追求“效率”的经验模型 Vina的设计哲学从一开始就偏向虚拟筛选——即在短时间内处理成千上万个分子。为此,它巧妙地“舍弃”了计算最耗时的静电和溶剂化项,转而用几个经验性的、拟合出来的高斯函数来近似描述范德华力和氢键作用。它独特的疏水项也能很好地捕捉疏水效应。这套组合拳让Vina在保持高精度的同时,速度极快,成为目前应用最广泛的对接软件之一
调用ad4力场
prepare_gpf.py -l ligand.pdbqt -r proH_rigid.pdbqt -y
autogrid4 -p proH_rigid.gpf -l proH_rigid.glg
vina --flex proH_flex.pdbqt \
--ligand ligand.pdbqt \
--maps proH_rigid \
--scoring ad4 \
--exhaustiveness 32 \
--out out_ad4.pdbqt生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号