Nat. Mach. Intell. | TDFold:用扩散模型突破AI单序列蛋白质结构预测瓶颈

Nat. Mach. Intell. | TDFold:用扩散模型突破AI单序列蛋白质结构预测瓶颈

DrugIntel
发布于 2026-04-13 15:10:42
发布于 2026-04-13 15:10:42

论文全称:Two-dimensional Geometric Template Diffusion for Boosting Single-Sequence Protein Structure Prediction 发表期刊:Nature Machine Intelligence(2026) DOI:10.1038/s42256-026-01210-2 接收日期:2026 年 2 月 25 日 | 在线发表:2026 年 4 月 1 日 代码链接:https://doi.org/10.5281/zenodo.18454943
一、研究背景与动机
1.1 蛋白质结构预测的两条技术路线
近年来,AI 驱动的蛋白质结构预测取得了里程碑式的进展。当前主流方法可归为两大范式:
同源依赖范式(Homology-based Methods)
以 AlphaFold2、AlphaFold3、RoseTTAFold 为代表。这类方法需要从 UniRef、PDB 等生物数据库中检索多序列比对(Multiple Sequence Alignment, MSA)和三维结构模板(3D structural template),并将其作为核心输入特征。其核心假设是:同源序列的共进化信息蕴含了蛋白质折叠的约束。
蛋白质语言模型范式(PLM-based Methods)
以 ESMFold、OmegaFold、RGN2、trRosettaX-single 为代表。这类方法完全基于氨基酸序列的文本信息,无需同源检索,可显著加速特征提取流程。然而,这些模型普遍采用深度堆叠的 Transformer 架构,以 ESMFold 为例,其 Evoformer 中的三角注意力机制(triangular attention)的时间复杂度为 O(n³),在长序列蛋白上面临严重的内存和计算瓶颈。
1.2 现有方法的两大核心痛点
痛点一:同源依赖导致泛化性差
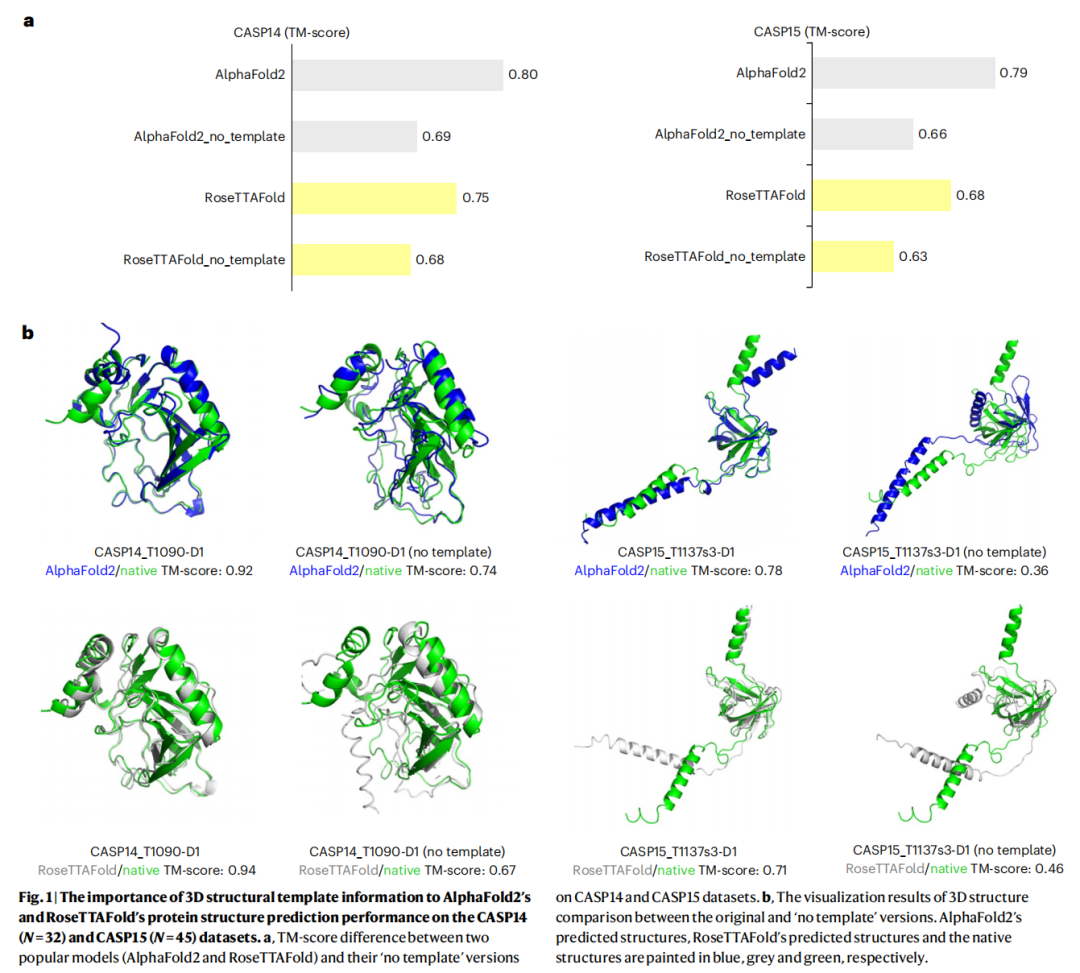
同源依赖方法对「孤儿蛋白」(Orphan proteins,即在数据库中缺乏同源序列的蛋白质)及快速演化的病毒蛋白预测效果极差。论文实验数据显示,在 CASP14 数据集上,移除三维结构模板后:
- • AlphaFold2 的 TM-score 从 0.80 → 0.69(下降 13.75%)
- • RoseTTAFold 的 TM-score 从 0.75 → 0.68(下降 9.33%)
在 CASP15 数据集上,情况更为严峻:
- • AlphaFold2 的 TM-score 从 0.79 → 0.66(下降 16.46%)
- • RoseTTAFold 的 TM-score 从 0.68 → 0.63(下降 7.35%)
对于真正缺乏同源信息的孤儿蛋白(Orphan 数据集),AlphaFold2(全同源模式)的 TM-score 仅为 0.37,AlphaFold3 为 0.41,远低于其在 CASP 数据集上的表现。
痛点二:PLM 方法计算成本居高不下
以 ESMFold 为代表的 PLM 方法,虽然无需同源检索,但其显存需求高达 8—20 GB(随序列长度线性—超线性增长),且推理时间随序列长度呈近立方级增长,对于含 500 个残基的蛋白质约需 100 秒。这使得大规模蛋白质结构预测任务,以及资源受限的学术机构的使用,都面临较大障碍。
1.3 核心研究问题
能否在不依赖同源数据库的前提下,同时实现高精度、低资源消耗、快速推理的单序列蛋白质结构预测?
二、方法:TDFold 整体框架
TDFold(Two-Dimensional Geometric Template Diffusion Fold)提出了一种端到端的两阶段网络架构,将视觉生成模型引入蛋白质结构预测领域。
氨基酸序列(单序列输入)
│
▼
┌─────────────────────────────────┐
│ 阶段一:2D 几何模板扩散模块 │
│ · 序列 → 文本提示词(Text LoRA) │
│ · SD 模型(冻结)+ UNet LoRA │
│ · 输出:残基间几何矩阵图像 │
│ - 距离矩阵 dC(Cβ–Cβ) │
│ - 方向矩阵 ω、θ、ϕ │
└─────────────────────────────────┘
│
▼(2D 几何模板)
┌─────────────────────────────────┐
│ 阶段二:序列-几何协同学习(SCL) │
│ · 残基级图学习分支 │
│ · 原子级图学习分支 │
│ · 变分融合框架 │
│ · SE(3)-EGNN 全原子坐标预测 │
└─────────────────────────────────┘
│
▼
三维蛋白质结构(全原子坐标)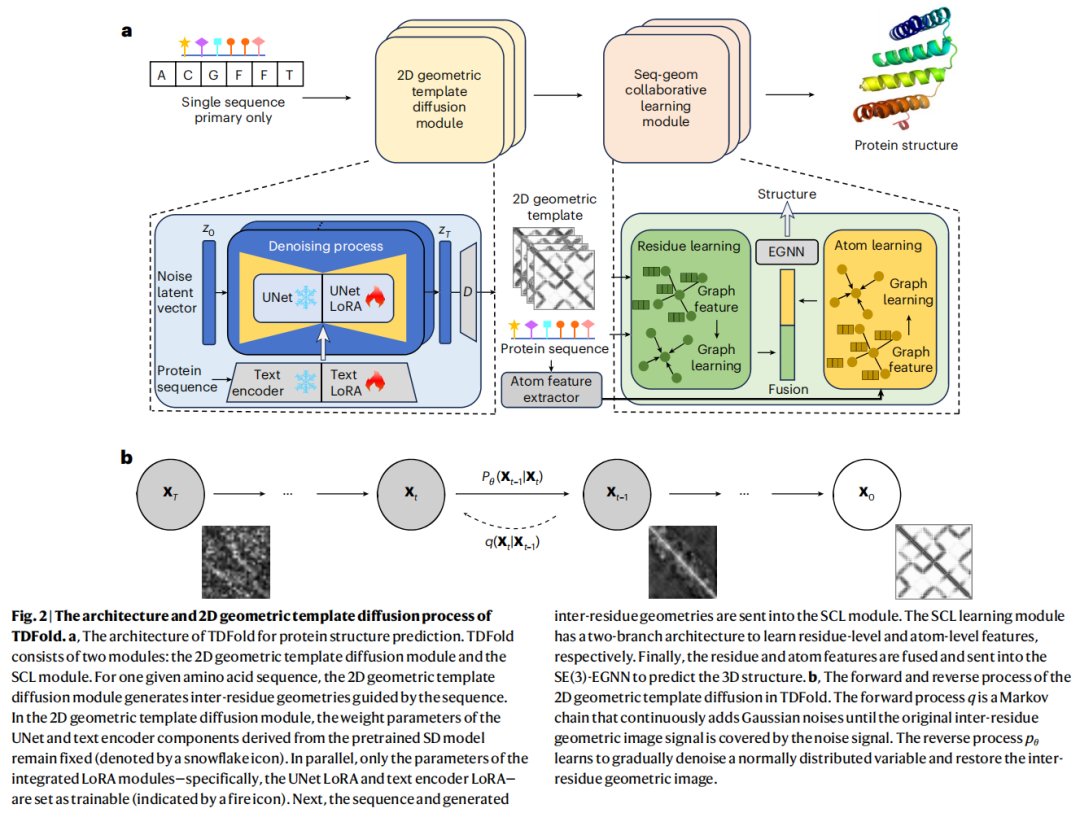
三大核心优势
优势维度 | TDFold | ESMFold | AlphaFold2 |
|---|---|---|---|
单序列预测精度 | ★★★★★ | ★★★★ | ★★(无模板模式) |
GPU 显存(500残基) | ~7 GB | ~20 GB | ~12 GB |
推理时间(500残基) | ~10 s | ~100 s | ~1000 s |
训练资源需求 | 单卡 4090,约1周 | 大规模集群 | 大规模集群 |
孤儿蛋白支持 | 优秀 | 一般 | 差 |
三、关键技术详解
3.1 第一阶段:二维几何模板扩散模块
3.1.1 核心洞见:几何矩阵即图像
论文的核心创新在于一个关键观察:残基间几何信息(inter-residue geometries)可以被编码为多通道的类图像特征矩阵。
具体而言,对于一个含 N 个残基的蛋白质,TDFold 定义了以下四种几何描述子:
几何量 | 物理含义 | 矩阵维度 | 离散化区间数 |
|---|---|---|---|
(Cβ–Cβ距离) | 残基对之间的空间距离 | N×N | 36(2.5Å–20.5Å,步长0.5Å) |
(Cα–Cβ–Cβ–Cα二面角) | 残基相对旋转 | N×N | 36(0°–360°) |
(N–Cα–Cβ–Cβ二面角) | 骨架方向 | N×N | 36(0°–360°) |
(Cα–Cβ–Cβ平面角) | 残基对平面取向 | N×N | 18(0°–180°) |
3.1.2 数据类型对齐:连续几何值 → RGB 图像像素值
由于 SD 模型要求输入为离散像素值(0–255),而几何量为连续值,需进行数据类型转换(Algorithm 1):
距离转换:
角度转换:
像素映射: 的矩阵值乘以 7(范围扩展至 0–252); 乘以 14(同样映射至 0–252)。最后,每种几何量的矩阵被复制为三通道,生成 RGB 图像,可直接输入 SD 模型处理。
3.1.3 稳定扩散模型与 LoRA 微调
为什么选择 SD 模型?
Stable Diffusion 具备强大的文本-图像协同建模能力,可以学习文本提示(氨基酸序列)与多通道矩阵(几何图像)之间的复杂分布映射。然而,SD 模型原本在通用图像-文本对上训练,直接应用于蛋白质几何生成存在两个鸿沟:
- • 数据类型鸿沟:连续几何值 vs 离散图像像素
- • 语义鸿沟:蛋白质序列语义 vs 自然语言描述图像的语义
分层 LoRA 微调策略
为弥合上述鸿沟,TDFold 对 SD 模型的两个关键组件分别引入 LoRA 分支:
Text LoRA(文本编码器适配)
原始 SD 的文本编码器(CLIP 编码器)支持最多 77 个 token,远不够描述典型蛋白质序列(可达数百至数千个残基)。TDFold 引入**位置插值(Position Interpolation)**扩展上下文窗口至 385 token:
对于更长的序列,采用非重叠分段策略逐段生成嵌入后聚合。
Text LoRA 的对齐损失函数为:
其目标是将蛋白质序列特征与几何图像特征映射到共享潜空间中。
UNet LoRA(去噪网络适配)
UNet LoRA 学习残基间几何数据的分布,并在蛋白质序列条件的引导下进行去噪生成:
UNet LoRA 的训练目标为:
冻结策略的重要性
SD 模型的原始参数完全冻结,仅训练 LoRA 参数。通过充分利用 SD 模型从 20 亿图像-文本对中习得的强大先验知识,LoRA 微调的泛化效果显著优于从头训练(消融实验中,LoRA 微调版在 CASP15 上 TM-score 提升 0.33,在 Orphan 上提升 0.21)。
推理加速:利用 DPMsolver、UniPC、DDIM 等高效采样器,去噪步数可从 1,000 步压缩至 25–50 步,大幅加速推理。
3.2 第二阶段:序列-几何协同学习(SCL)模块
SCL 模块是一个轻量级图神经网络,包含四个核心组件。
3.2.1 残基级图学习(Residue-level Graph Learning)
该分支学习序列与几何的交互表示,包含三个子网络:
① 协同学习网络
对蛋白质序列(tokenize 后嵌入为特征向量)和残基间几何对(从生成的几何矩阵中提取距离和相对方向)进行双向迭代更新:
- • 外积(outer product):序列特征图计算外积后累加至对特征,将序列信号注入几何对表示
- • 交叉注意力(cross-attention):从几何对到序列的注意力传递,将结构信息反馈至序列表示
- • 自注意力层:对序列和对特征分别进行自我精化
物理对称性保证:对 (Cβ–Cβ 距离)和 二面角矩阵执行对称化操作 ,显式保证其物理对称性。
② 混合卷积神经网络(Hybrid CNN)
采用非对称与对称两类卷积核学习残基间几何特征:
- • 非对称核:捕获具有方向性的几何量(二面角 和平面角 )
- • 对称核:建模天然对称的几何量(距离 和二面角 )
输出残基级关联矩阵 ,作为图网络的边特征。
③ 残基级图神经网络
构建残基图(残基为节点,边编码 CNN 精化后的几何关系),采用图 Transformer 架构进行表示学习:
3.2.2 原子级图学习(Atom-level Graph Learning)
为显式建模侧链对骨架构象的影响,引入原子级图分支:
- • 为 20 种标准氨基酸分别构建图表示(原子为节点,共价键为边)
- • 通过模拟脱水反应(水分子消除形成肽键 –CO–NH–),将氨基酸图组装为完整蛋白质原子图
- • 使用三层 GNN 模块学习原子表示,并通过残差连接增强表达能力
3.2.3 残基-原子特征融合(Variational Fusion)
设计了一套双层次交互学习的变分融合框架:
- 1. 相关矩阵计算:,度量第 个残基与第 个原子的相关性
- 2. Bernoulli 概率采样:引入隐变量 ,每个元素 表示第 个原子影响第 个残基折叠的概率,通过后验分布 学习,过滤冗余连接
- 3. 特征投影与拼接:将筛选后的原子特征投影至残基特征空间(),与残基特征拼接形成残基-原子交互表示
3.2.4 SE(3)-EGNN 全原子坐标预测
最终的三维结构预测分两个阶段:
骨架预测:
- • 用 MLP 从融合特征投影出初始骨架坐标
- • 构建残基图(连接 top-k 空间近邻和序列相邻残基)
- • 通过 SE(3)-等变图神经网络(SE(3)-EGNN)精化骨架坐标
- • 利用球谐函数和 Clebsch–Gordan 展开保证 SE(3) 等变性,确保旋转/平移变换下输出的一致性
全原子精化:
- • 将侧链原子附加至精化后的骨架
- • 以分子动力学原理为启发,通过朗之万积分器(Langevin Integrator)引入轻微布朗运动,优化侧链构象(骨架固定),防止空间碰撞
3.2.5 损失函数
SCL 模块的训练损失为多任务联合目标:
其中 包含键长、键角和二面角的 RMSE 约束。
全原子训练阶段在骨架损失基础上加入侧链约束:
四、实验评估
4.1 数据集设置
训练集:来自 PDB 的 2020 年 5 月 1 日前发布的 352,409 个非冗余蛋白质结构域。
测试集(五个基准):
数据集 | 蛋白质数量 | 特点 |
|---|---|---|
Orphan | 77 | 在 UniRef30、PDB70、MGnify 中均无同源序列 |
Orphan25 | 25 | 搜索 UniRef50_2018_03 数据库返回零同源序列,PDB 2020年5月后发布 |
CASP14 | 32 | 标准竞赛基准 |
CASP15 | 45 | 标准竞赛基准 |
CASP16 | 15 | 最新竞赛基准 |
数据无泄露保证:训练集与所有测试集之间的结构相似性(TM-score)均低于 0.3,序列一致性(BLAST)均低于 30%。
4.2 孤儿蛋白结构预测(Orphan & Orphan25)
在最具挑战性的孤儿蛋白数据集上,TDFold 全面超越所有对比方法:
Orphan 数据集(N=77)
方法 | TM-score ↑ | GDT_TS ↑ | pLDDT ↑ | 输入模式 |
|---|---|---|---|---|
TDFold | 0.46 | 77.50 | 71.85 | 单序列 |
ESMFold | 0.42 | 72.08 | 69.52 | 单序列 |
OmegaFold | 0.39* | 74.44 | 68.75 | 单序列 |
RGN2 | — | — | — | 单序列 |
AlphaFold3 | 0.41 | 73.49 | 69.55 | 全同源 |
AlphaFold2 | 0.37 | 68.91 | 67.23 | 全同源 |
RoseTTAFold | 0.35 | 67.61 | 65.96 | 全同源 |
*OmegaFold 与 TDFold 对比 p=0.0227
Orphan25 数据集(N=25)
方法 | TM-score ↑ | GDT_TS ↑ | pLDDT ↑ | 输入模式 |
|---|---|---|---|---|
TDFold | 0.61 | 68.37 | 67.48 | 单序列 |
ESMFold | 0.54 | 64.93 | 64.65 | 单序列 |
OmegaFold | 0.52* | 63.65 | 54.25 | 单序列 |
AlphaFold3 | 0.57 | 65.46 | 65.57 | 全同源 |
AlphaFold2 | 0.44 | 61.70 | 64.87 | 全同源 |
RoseTTAFold | 0.40 | 58.25 | 63.25 | 全同源 |
*OmegaFold 与 TDFold 对比 p=0.0256
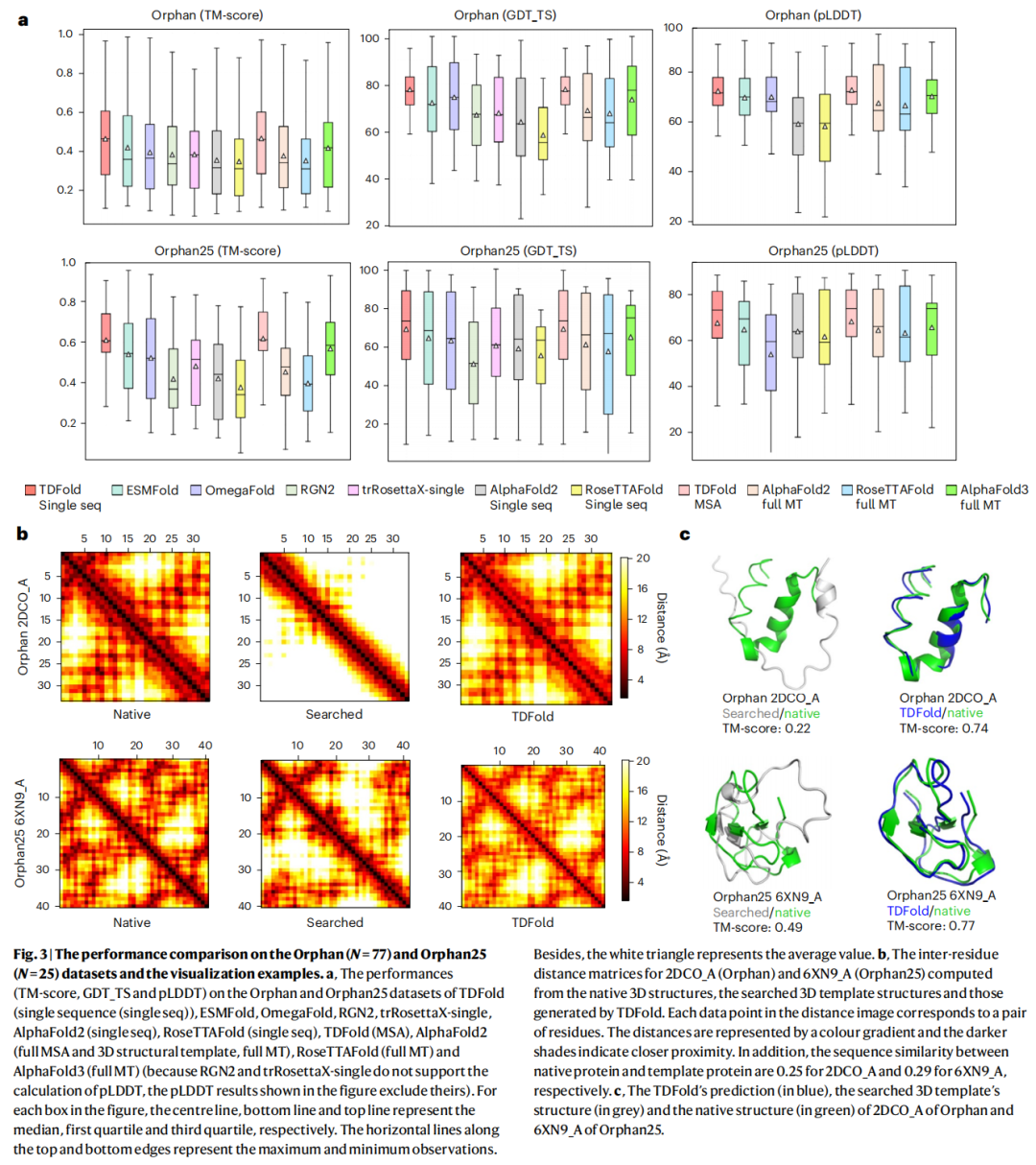
关键发现:TDFold 在单序列模式下的 TM-score(0.46/0.61),不仅远超其他单序列方法,甚至超过了使用完整 MSA 和三维结构模板的 AlphaFold2(0.37/0.44)和 AlphaFold3(0.41/0.57)。这验证了 2D 几何模板扩散路线在同源信息缺乏时的核心价值。
4.3 通用蛋白质结构预测(CASP14/15/16)
三大竞赛基准综合表现
数据集 | TDFold | ESMFold | OmegaFold | AlphaFold2(全同源) | AlphaFold3(全同源) |
|---|---|---|---|---|---|
CASP14 TM | 0.73 | 0.71 | 0.76 | 0.80 | — |
CASP15 TM | 0.70 | 0.69 | 0.63* | 0.79 | — |
CASP16 TM | 0.77 | 0.75 | 0.69* | 0.78 | — |
*CASP15 OmegaFold p=0.0268;CASP16 OmegaFold p=0.0246
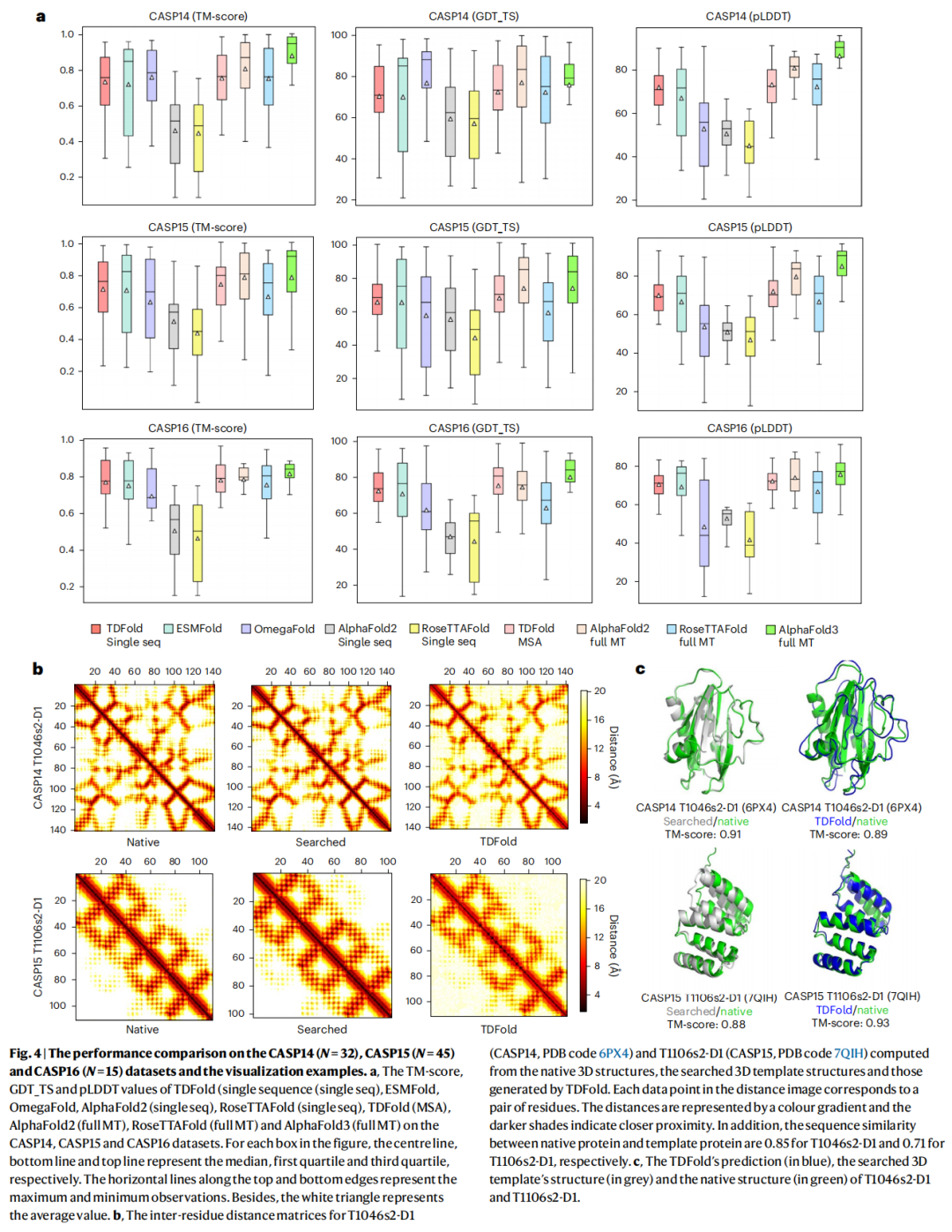
说明:TDFold 在单序列模式下持续超越 ESMFold,且在 CASP15 和 CASP16 上显著优于 OmegaFold。与使用全同源信息的 AlphaFold2/AlphaFold3 的差距也明显缩小。
OmegaFold 在 CASP14 上(0.76)略高于 TDFold(0.73)的合理解释:OmegaFold 的训练集包含 2021 年 4 月前的 UniRef50 序列和 PDB 结构,而 CASP14 目标蛋白于 2020 年 7 月起陆续释放至 PDB,存在一定数据泄露风险;TDFold 及其他方法的训练截止日期均为 2020 年 5 月,不存在此问题。
4.4 病毒蛋白结构预测
针对同源序列极少(< 20 条)的快速演化病毒蛋白:
目标蛋白 | 病毒类型 | 序列长度 | MSA 数量 | AlphaFold2 | AlphaFold3 | ESMFold | TDFold |
|---|---|---|---|---|---|---|---|
T1033-D1 | crAs 噬菌体 | 100 | 3 | 0.41 | 0.44 | 0.33 | 0.94 |
T1039-D1 | crAs 噬菌体 | 161 | 3 | 0.58 | 0.47 | 0.28 | 0.89 |
T1064-D1 | SARS-CoV-2 ORF8 | 75 | 14 | 0.41 | 0.71 | 0.44 | 0.73 |
T1082-D1 | T4 噬菌体 | 71 | 11 | 0.40 | 0.73 | 0.38 | 0.76 |
T1099-D1 | 肝炎病毒 | 178 | 10 | 0.80 | 0.85 | 0.47 | 0.87 |
T1123-D1 | 星状病毒 | 214 | 9 | 0.62 | 0.28 | 0.31 | 0.79 |
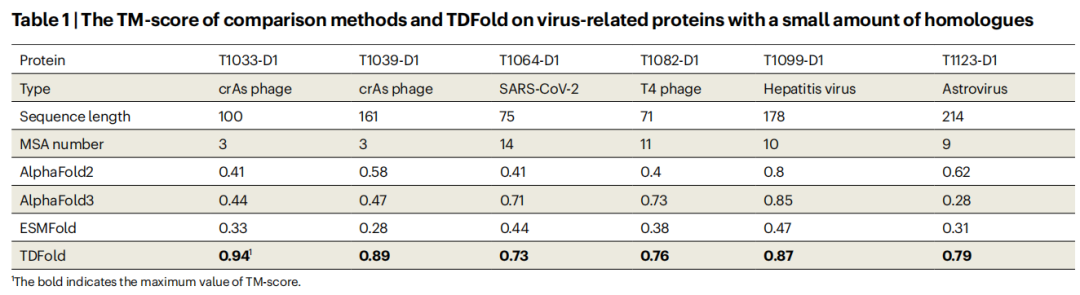
TDFold 在所有六个病毒蛋白靶点上均取得最高 TM-score,充分展示了其在同源信息极度稀缺情境下的强大泛化能力。
4.5 推理效率对比
推理时间(以蛋白质序列长度为变量,N=194 个靶点):
序列长度范围 | TDFold | ESMFold | AlphaFold3 | AlphaFold2 / RoseTTAFold |
|---|---|---|---|---|
≤ 140 残基 | 略高于 ESMFold | 最快 | — | 最慢 |
140–500 残基 | 最快 | 约 100 s | 约 240 s | > 1000 s |
> 500 残基 | ~10 s(稳定) | 持续增加 | 持续增加 | 持续增加 |
关键特性:TDFold 的推理时间几乎不随序列长度增加,因为其主要由去噪步数决定,而非序列长度。这一特性使其在长序列蛋白的大规模预测任务中具有决定性优势。
GPU 显存占用(随序列长度增加的变化幅度):
方法 | 基础显存 | 最大显存(500残基) | 增幅 |
|---|---|---|---|
TDFold | ~5 GB | ~7 GB | +40% |
AlphaFold2 | ~4 GB | ~12 GB | +200% |
RoseTTAFold | ~5 GB | ~16 GB | +220% |
ESMFold | ~8 GB | ~20 GB | +150% |
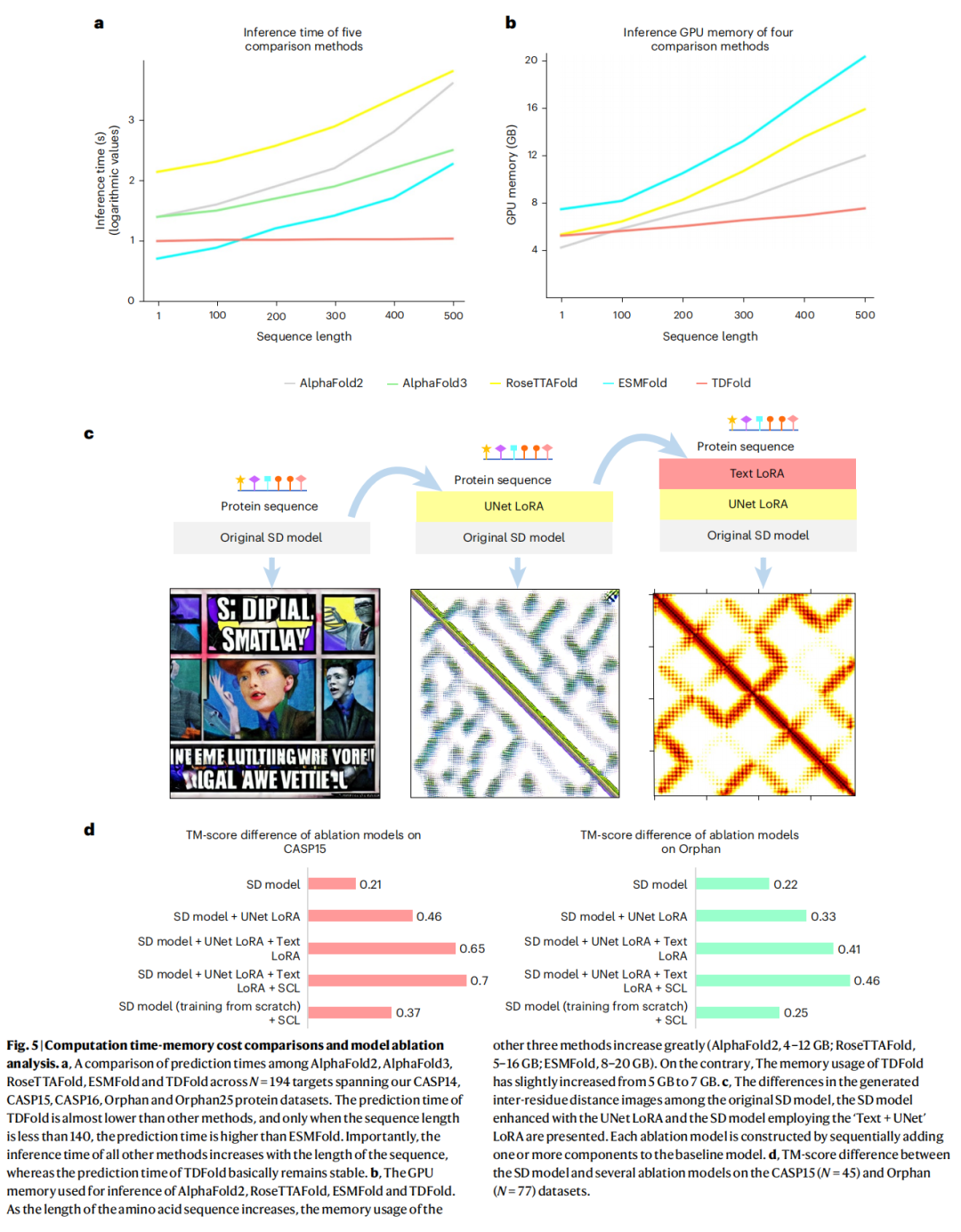
4.6 几何质量评估
论文采用 KL 散度(Kullback–Leibler divergence)定量评估生成几何模板与真实值之间的分布差异,并与 trRosetta 的几何预测结果对比。在 Orphan、Orphan25 以及所有 CASP 数据集上,TDFold 生成的几何模板的 KL 散度均低于 trRosetta,说明 TDFold 生成的几何模板质量更优。
可视化案例:在 2DCO_A(Orphan)和 6XN9_A(Orphan25)两个典型案例中:
- • 数据库检索到的结构模板(序列相似度分别仅为 0.25 和 0.29)与真实结构差异显著
- • TDFold 生成的距离矩阵与真实矩阵高度吻合
- • 使用 TDFold 生成几何的预测 TM-score(0.74、0.77)远高于使用检索模板的结果(0.22、0.49)
五、消融实验
逐步叠加各模块,在 CASP15(N=45)和 Orphan(N=77)数据集上评估各组件贡献:
模型配置 | CASP15 TM-score | Orphan TM-score |
|---|---|---|
原始 SD 模型(基线) | 0.21 | 0.22 |
+ UNet LoRA | 0.46 (+0.25) | 0.33 (+0.11) |
+ Text LoRA | 0.65 (+0.19) | 0.41 (+0.08) |
+ SCL 模块 | 0.70 (+0.05) | 0.46 (+0.05) |
SD(从头训练)+ SCL | 0.37 | 0.25 |
关键结论:
- 1. UNet LoRA 贡献最大(CASP15 +0.25,Orphan +0.11),是驱动几何模板生成的核心组件
- 2. Text LoRA 同样不可或缺(CASP15 +0.19,Orphan +0.08),确保序列嵌入与几何图像嵌入的语义对齐
- 3. SCL 模块 在几何质量保证的基础上,进一步提升结构预测精度(约 +0.05)
- 4. 预训练 LoRA 微调 vs 从头训练:LoRA 微调版在 CASP15 上高出 0.33,在 Orphan 上高出 0.21,充分验证了迁移 SD 模型强大先验知识的重要性
可视化佐证:仅使用原始 SD 模型时,以蛋白质序列为提示词生成的「图像」呈现为含有字符和人物的通用图像,说明模型完全无法理解蛋白质序列的语义;加入 UNet LoRA 后,生成图像开始出现对角线结构;同时加入 Text LoRA 后,才能生成与真实几何结构高度吻合的残基间距离图像。
六、讨论与局限性
6.1 方法贡献总结
- 1. 技术路线创新:首次将视觉生成扩散模型(Stable Diffusion)系统性引入蛋白质结构预测,开辟了「以文生几何图像」的新范式
- 2. 同源信息解耦:通过生成高质量几何模板,完全绕开同源数据库检索,在孤儿蛋白预测上取得最优性能
- 3. 效率-精度双优:在单序列模式下超越 ESMFold 的同时,将显存需求降低约 65%,推理速度提升 10×(相比 ESMFold)至 100×(相比 AlphaFold2)
- 4. 资源门槛降低:可在单张消费级 GPU(NVIDIA 4090 24GB)上完成完整训练,显著降低了高质量蛋白质结构预测的计算资源门槛
6.2 潜在局限与未来方向
- • CASP 数据集上与全同源方法仍有差距:在拥有丰富同源信息的场景下,AlphaFold2/AlphaFold3 依然保持优势(如 CASP14 AlphaFold2 TM=0.80 vs TDFold 0.73);TDFold 的优势主要体现在同源信息匮乏的场景
- • CASP16 样本量较小:仅 15 个结构,统计结论需谨慎解读
- • 几何模板的多样性:扩散模型的随机性可能导致不同运行结果存在差异,多次采样并集成可能进一步提升性能
- • 蛋白质复合物与大分子:当前工作聚焦于单链蛋白质,向蛋白质复合物的扩展是自然的后续方向
七、总结
TDFold 代表了蛋白质结构预测领域的一次方法论突破:通过将视觉扩散生成模型迁移应用于蛋白质几何特征生成,它在不依赖同源信息的前提下,实现了对 ESMFold 等主流 PLM 方法的全面超越,并在孤儿蛋白和病毒蛋白等高难度任务上展现出尤为突出的优势。
更值得关注的是,TDFold 将高质量蛋白质结构预测从大型计算集群「带回」到了普通科研工作站,这对推动全球资源受限机构的蛋白质科学研究具有深远意义。随着扩散模型技术的持续发展,这一技术路线有望在蛋白质功能预测、药物靶点识别、蛋白质设计等下游任务中得到更广泛的应用。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号