AlphaFold 数据库重磅扩展:蛋白质复合物结构预测的规模化突破

AlphaFold 数据库重磅扩展:蛋白质复合物结构预测的规模化突破

DrugIntel
发布于 2026-04-13 15:12:20
发布于 2026-04-13 15:12:20

文献来源: Han Y, Tsenkov MI, Venanzi NAE, et al. AlphaFold Database expands to proteome-scale quaternary structures. bioRxiv (2026). https://doi.org/10.64898/2026.03.27.714458 预印本发布时间: 2026年3月29日 通讯作者: Sameer Velankar(EMBL-EBI)、Martin Steinegger(首尔国立大学)、Jennifer Fleming(EMBL-EBI)、Milot Mirdita(成均馆大学)、Christian Dallago(NVIDIA)
导读
AlphaFold2 的问世开创了蛋白质单体三维结构预测的新时代,使研究者得以以前所未有的规模获取蛋白质结构信息。然而,生物学现实是:蛋白质极少以孤立单体的形式发挥功能,绝大多数细胞过程由蛋白质复合物——即蛋白质四级结构——所驱动。蛋白质-蛋白质相互作用(PPI)界面是信号传导、基因表达调控、酶催化、免疫识别等几乎所有核心生物过程的结构基础,同时也是药物设计的重要靶点。
本文介绍的工作,由首尔国立大学、EMBL-EBI、NVIDIA 与 Google DeepMind 等机构联合完成,是迄今为止规模最大的蛋白质复合物结构预测研究,将 AlphaFold 蛋白质结构数据库(AFDB)从单体扩展至蛋白质组规模的四级结构,系统性地弥补了结构互作组(structural interactome)的覆盖空白。
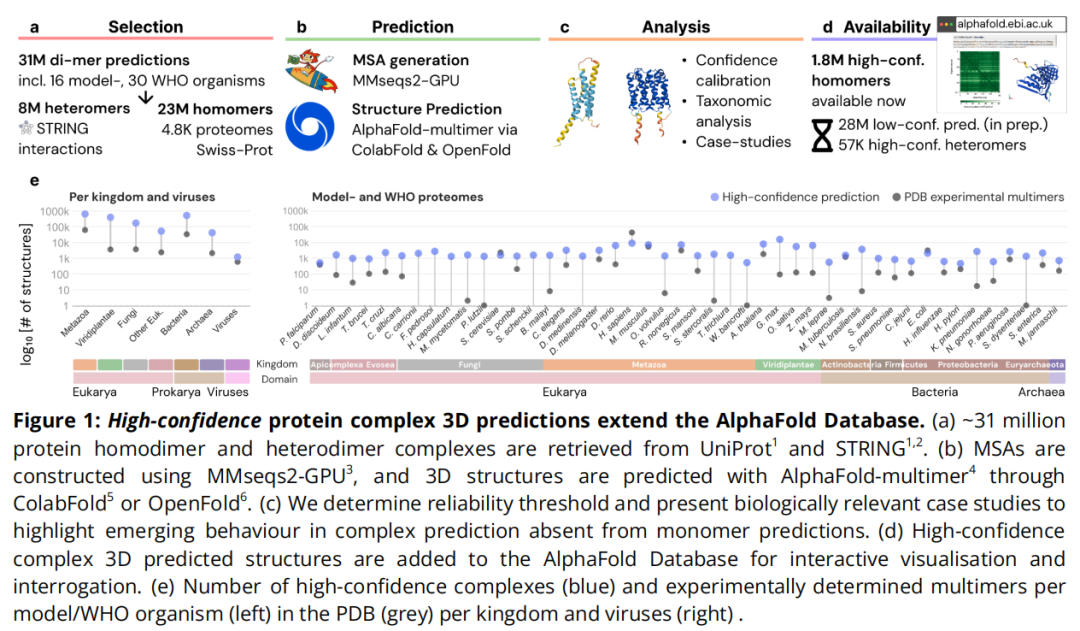
一、研究背景与问题定义
1.1 现有结构数据库的覆盖局限
蛋白质数据库(PDB)是存储实验测定结构的权威数据库,但其对蛋白质-蛋白质相互作用界面的覆盖极为有限。大量已在 STRING、IntAct 等数据库中记录的相互作用,缺乏对应的三维结构信息。这一结构空白对以下领域构成根本性瓶颈:
- • 功能机制研究:许多蛋白质的构象变化、变构调控、酶活位点等仅在复合物状态下方能被准确描述;
- • 药物设计:以 PPI 界面为靶标的小分子或生物制品设计,严重依赖界面结构信息;
- • AI 模型训练:蛋白质设计与功能预测模型需要大量高质量的复合物结构作为训练数据。
1.2 已有计算预测工作的不足
尽管 AlphaFold-Multimer(2021)、RoseTTAFold 等方法已证明高置信度复合物预测的可行性,但此前的蛋白质组规模研究普遍存在以下问题:
- • 覆盖生物体有限,通常仅针对特定模式生物;
- • 缺乏系统性的置信度校准标准,难以跨研究比较;
- • 预测结果未整合入稳定的公共数据库基础设施,复用性差;
- • 计算规模受硬件限制,无法覆盖全球健康相关物种。
二、研究设计与数据规模
2.1 预测范围
类型 | 数量 | 来源 |
|---|---|---|
同源二聚体(homodimers) | 23,441,822 | UniProt 4,777 个蛋白质组(含 Swiss-Prot) |
异源二聚体(heterodimers) | 7,620,644 | STRING 物理相互作用注释 |
总计 | ~31,062,466 | — |
蛋白质组覆盖范围包括:
- • 16 种模式生物(如人、小鼠、酵母、果蝇、秀丽隐杆线虫、大肠杆菌等)
- • 30 个 WHO 全球健康重点蛋白质组(涵盖结核、疟疾、血吸虫病、锥虫病等被忽视热带病相关病原体)
- • 4,777 个 UniProt 参考蛋白质组
序列长度限制:单体 15–1,500 个氨基酸;复合物最大 3,000 个氨基酸(两链之和)。
2.2 异源二聚体的获取策略
异源二聚体候选对从 STRING v12.0 物理相互作用数据集提取。研究者刻意不设置 STRING 分值阈值,以最大化覆盖度,确保对重点蛋白质组的无偏探索——这与多数此前研究的策略不同,后者通常在 STRING score > 500 或 > 700 处截断。
STRING ID 到 UniProt 登录号的映射采用三步级联策略:
- 1. UniProt 官方 ID 映射(最高优先级)
- 2. CRC64 序列哈希匹配
- 3. MMseqs2 精确序列比对(100% 序列一致性,≥95% 双向覆盖度)
最终映射率:模式生物集 73.1%,WHO 健康蛋白质组集 82.4%,去冗余后获得 7,620,644 个候选异源二聚体。
三、计算架构与工程实现
3.1 MSA 生成
使用 MMseqs2-GPU(ColabFold 1.6.0 预发布版)生成多序列比对(MSA):
- • 数据库:UniRef30(2302版本),提供 UniRef100 的聚类覆盖;
- • 关键创新:实施了一种单分类群最优比对策略(best-hit-per-taxon filter),每个物种仅保留得分最高的同源序列,构成一种实用的直系同源过滤方案。该策略通过防止旁系同源序列稀释进化协同信号,显著提升复合物预测质量;
- • 异源 MSA 构建:直接拼接两条链的同源二聚体 MSA,不进行额外的序列配对(pairing)。研究者比较了基于物种的配对策略与简单拼接策略,发现在较高 ipTM 阈值(>0.8)下,简单拼接并不劣于配对,且大幅降低计算复杂度。
3.2 结构预测引擎
两套推理系统并行使用:
ColabFold(主用于同源二聚体)
- • 核心模型:AlphaFold-Multimer model_1_multimer_v3
- • 参数:4次循环迭代(recycling),启用早停,不进行弛豫(relaxation)
- • 优化:同长序列打包批处理(batch packing),按 MSA 深度降序排序,减少 JAX(v0.7.2)模型重新编译次数
- • 新增参数
--skip-output msa,plots,pae_json,只保留 .pdb 和 .json 文件,显著减少 I/O 开销
OpenFold(集成 NVIDIA TensorRT + cuEquivariance)
- • 无需 JAX 重新编译,天然支持异构批次
- • 针对长序列,将 CPU 特征化(featurization)与 GPU 推理并行执行
- • 已验证与 ColabFold 输出等价
3.3 高性能计算扩展
- • 部署于 NVIDIA DGX H100 超级节点,以节点为单位(每节点 8× H100 GPU)扩展
- • 采用 Slurm 任务阵列框架,每个任务独立处理约 5,000 个结构的分片
- • 每节点写入本地暂存,完成后通过 s5cmd 直接上传至对象存储,避免共享 Lustre 文件系统的 I/O 竞争
- • 双层断点续跑机制:阶段级跳过 + 分片级跳过,对节点故障和 Slurm 抢占具有鲁棒性
3.4 后处理工具链
开发了一套 AFDB 集成工具包(AFDB-Integration-Kit),涵盖:
- • 接口质量指标计算(ipSAE、ipTM、pDockQ2、LIS):ipSAE 通过 C++ 分片批处理实现,高吞吐;
- • 结构碰撞检测:GPU 加速脚本,每批打包最多 512 个结构,使用
torch_cluster.radius_graph执行原子距离核计算; - • 二级结构注释:PyDSSP(矢量化 NumPy/PyTorch 实现),整批处理,无子进程开销;
- • JSON 解析替换为 orjson(3.11.4),大型 PAE 矩阵反序列化速度提升约一个数量级;
- • PDB 解析替换为 GEMMI;元数据查询批量化为 DuckDB,按分片一次性执行,消除每个模型的数据库往返开销;
- • 数据聚合使用 PyArrow 批次流式处理,避免全数据集载入内存;
- • 生成符合 ModelCIF 标准的 mmCIF 和 Binary CIF 文件,直接用于 AFDB 数据库摄取。
四、置信度评估体系
4.1 指标体系
在模型默认输出的 pLDDT 和 ipTM 之外,研究团队额外计算了以下指标:
指标 | 全称 | 说明 |
|---|---|---|
ipSAE | Interface predicted Score from Aligned Errors | 基于 PAE 矩阵的界面置信度评分,具有方向性,分别计算 A→B 和 B→A |
ipSAEmin | — | 取 ipSAE(A→B) 和 ipSAE(B→A) 的较小值,作为保守估计 |
pDockQ2 | — | 基于界面残基接触的对接质量评分 |
LIS / LISmin | Local Interaction Score | 局部相互作用评分及其最小值 |
clashbackbone | — | 主链原子碰撞数 |
clashheavy-atom | — | 重原子碰撞数 |
4.2 置信度阈值的确定
验证数据集构建:
- • 正例:1,968 个 PDB 同源二聚体(2021年9月30日后发布,与 AlphaFold-Multimer 训练集分离,且与预测集序列精确匹配)
- • 负例:2,211 个 PDB 单体(预期在同源二聚体预测中得分低)
指标比较: 对 ipTM、ipSAEmin、LISmin、pDockQ2 四项指标分别计算 precision-recall-F1 曲线。ipSAEmin 表现出最清晰的分布分离和最稳定的 F1 平台期。
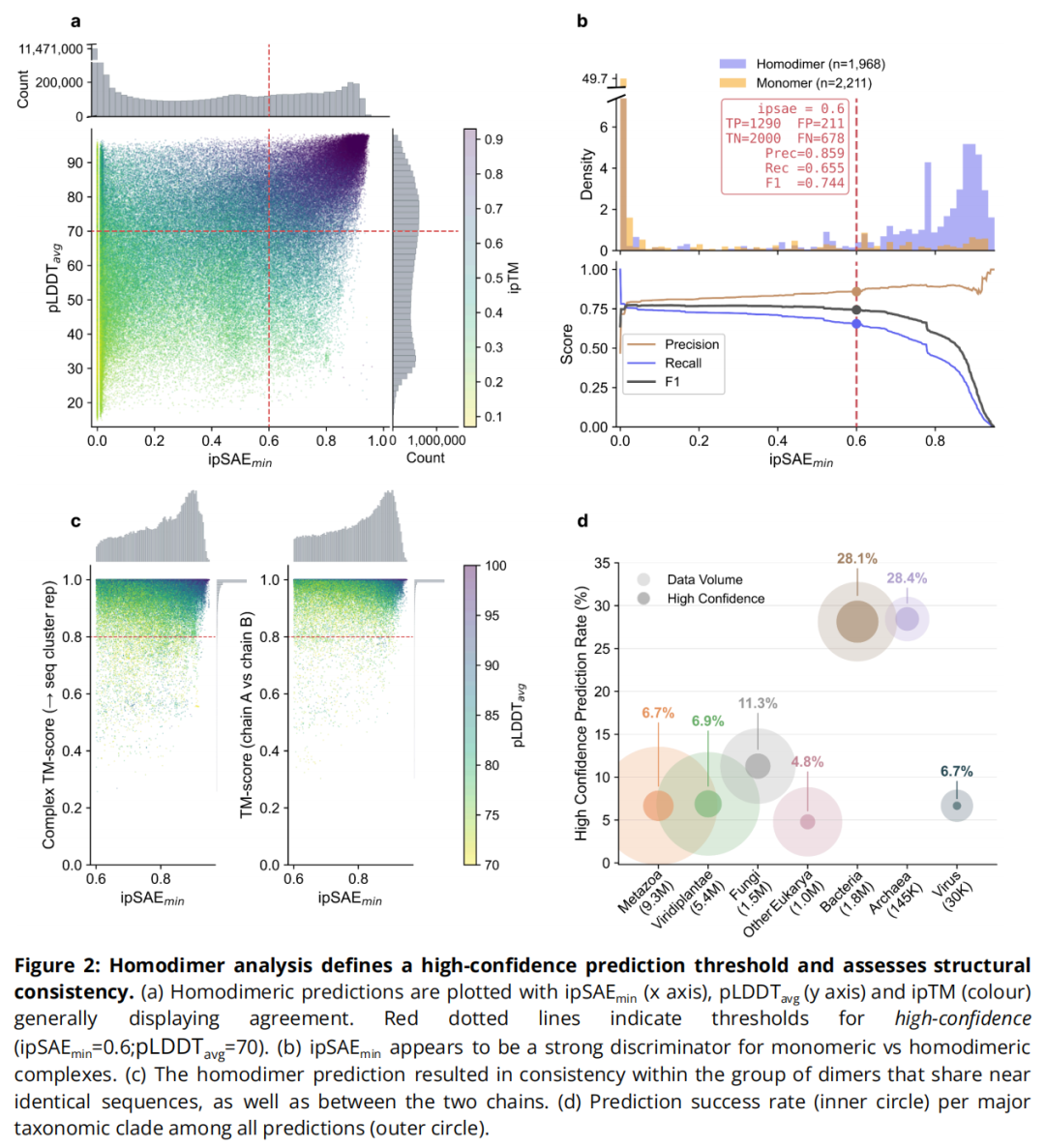
最终高置信度标准(三项联合):
在 ipSAEmin = 0.6 阈值处:精确率(Precision)= 0.859,召回率(Recall)= 0.655,F1 = 0.744
pLDDTavg ≥ 70 额外排除了约 15% 的数据,与 AFDB 单体高置信度标准保持一致。clash 过滤进一步去除空间冲突严重的模型。
4.3 AFDB 网站置信度分级
为方便非专业用户解读,AFDB 将同源二聚体按 ipSAEmin 进一步分为三个展示级别:
级别 | ipSAEmin 范围 | 解读建议 | 条目数 |
|---|---|---|---|
极高置信度(Very High Confidence) | ≥ 0.8 | 界面精度高,可直接用于机制分析 | 972,625 |
置信(Confident) | 0.7 – < 0.8 | 相互作用正确,界面解析良好概率高 | 438,879 |
低置信度(Low Confidence) | 0.6 – < 0.7 | 存在相互作用信号,建议谨慎解读 | 342,738 |
五、同源二聚体分析结果
5.1 整体覆盖与筛选
- • 输入:23,441,822 个同源二聚体预测
- • 高置信度输出:1,754,242 个(约 7% 保留率)
- • 已在 AFDB 上线,支持搜索、交互式可视化及批量下载
5.2 预测内部一致性验证
序列聚类内结构一致性:
对高置信度同源二聚体集,用 MMseqs2 以 98% 序列一致性、95% 覆盖度聚类,得到 1,429,305 个聚类(148,148 个非单例聚类,平均聚类大小 2.96)。将每个成员结构与聚类代表用 Foldseek Multimer 对齐:95.9% 的复合物达到查询归一化或靶标归一化的复合 TM-score > 0.8,证明数据集内部结构一致性良好。
同一二聚体两链间一致性:
用 Foldseek 对每个预测同源二聚体的 A、B 两链进行结构对齐:98.81% 的预测达到 TM-score > 0.8,进一步确认预测结果可靠。
5.3 分类群间预测成功率差异
分类群 | 高置信度比例 | 主要原因 |
|---|---|---|
古菌(Archaea) | ~28.4% | 蛋白质结构紧凑,同源寡聚体普遍 |
细菌(Bacteria) | ~28.1% | 同上 |
真菌(Fungi) | ~11.3% | 蛋白质较长,多结构域,无序区增多 |
后生动物(Metazoa) | ~6.7% | 多结构域、高度无序,更多异源复合物 |
绿色植物(Viridiplantae) | ~6.9% | 同上 |
病毒(Viruses) | ~6.7% | — |
古菌和细菌的成功率是后生动物的 3 倍以上,反映了真核蛋白质在进化过程中获得了更长、更复杂、更多无序区域的结构特征,且功能上更依赖异源复合物组装。
六、异源二聚体分析结果
6.1 预测结果与 STRING 分值的关系
对 7,620,644 个异源二聚体应用与同源二聚体相同的过滤标准,得到 56,956 个暂定高置信度预测。研究发现:
- • STRING 分值与高置信度预测率之间存在显著正相关(p < 10⁻³⁰⁰),OR = 1.44(逻辑回归),表明具有更多实验证据支持的相互作用确实更容易获得可信结构。
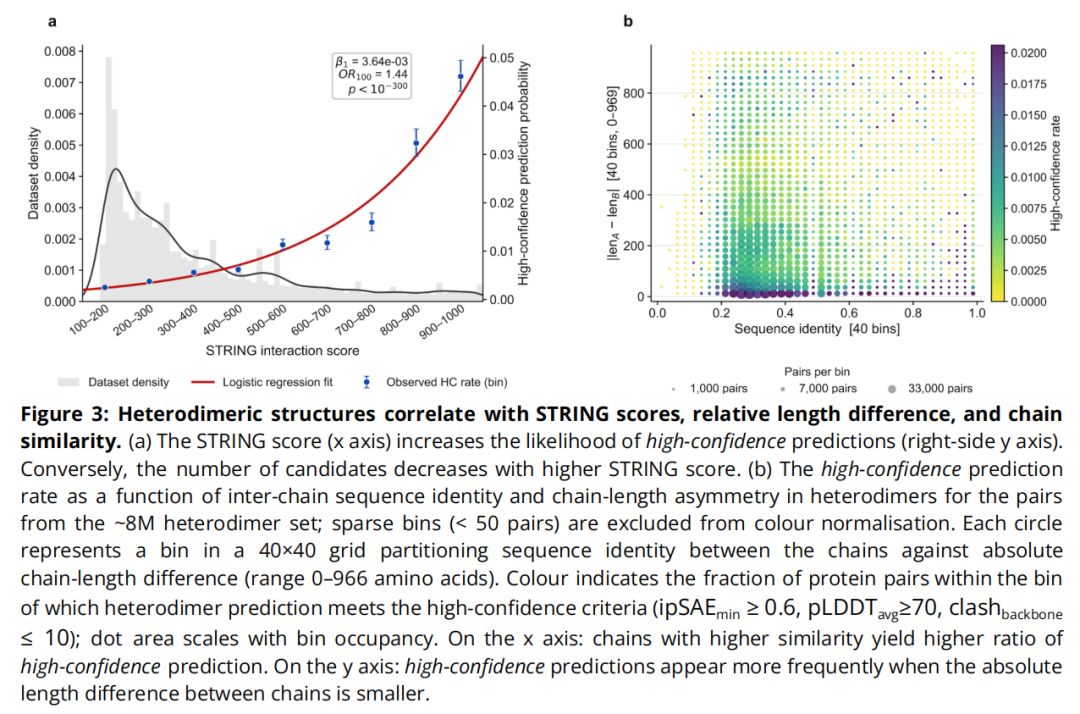
6.2 影响异源二聚体预测成功率的结构因素
链间序列相似性(x 轴): 序列一致性越高,高置信度预测率越高; 链长差异(y 轴): 两链绝对长度差越小,预测成功率越高。
两者效应独立存在:即便在低序列相似性区间(如 0.2–0.4),链长差异对预测成功率的负面影响依然显著。
关键局限: 当前过滤标准(源自同源二聚体校准)偏向于"类同源二聚体"特性(链间相似性高、长度对称),对典型异源二聚体(两链差异显著)存在系统性偏倚。作者因此将此批结构标注为"暂定高置信度",计划在后续版本中专门针对异源二聚体进行置信度重校准。
七、高置信度复合物空间的聚类分析
7.1 聚类方法
使用 Foldseek Multimercluster 对 1,811,201 个结构(1,754,242 同源 + 56,959 异源)进行聚类,参数:
- • 3Di 序列覆盖度 ≥ 60%(
-c 0.6) - • 界面 LDDT 阈值 ≥ 0.3(
--interface-lddt-threshold 0.3) - • 链 TM-score 阈值 ≥ 0.7(
--chain-tm-threshold 0.7)
7.2 聚类结果
- • 总聚类数:224,862
- • 非单例聚类数:86,975(包含至少 2 个成员)
- • 压缩比:约 8 倍
- • 同源二聚体主导:非单例条目中约 82% 为同源二聚体
- • 混合聚类(同时含同源与异源成员)仅占非单例聚类的 ~0.9%,主要见于最大的 10% 聚类中
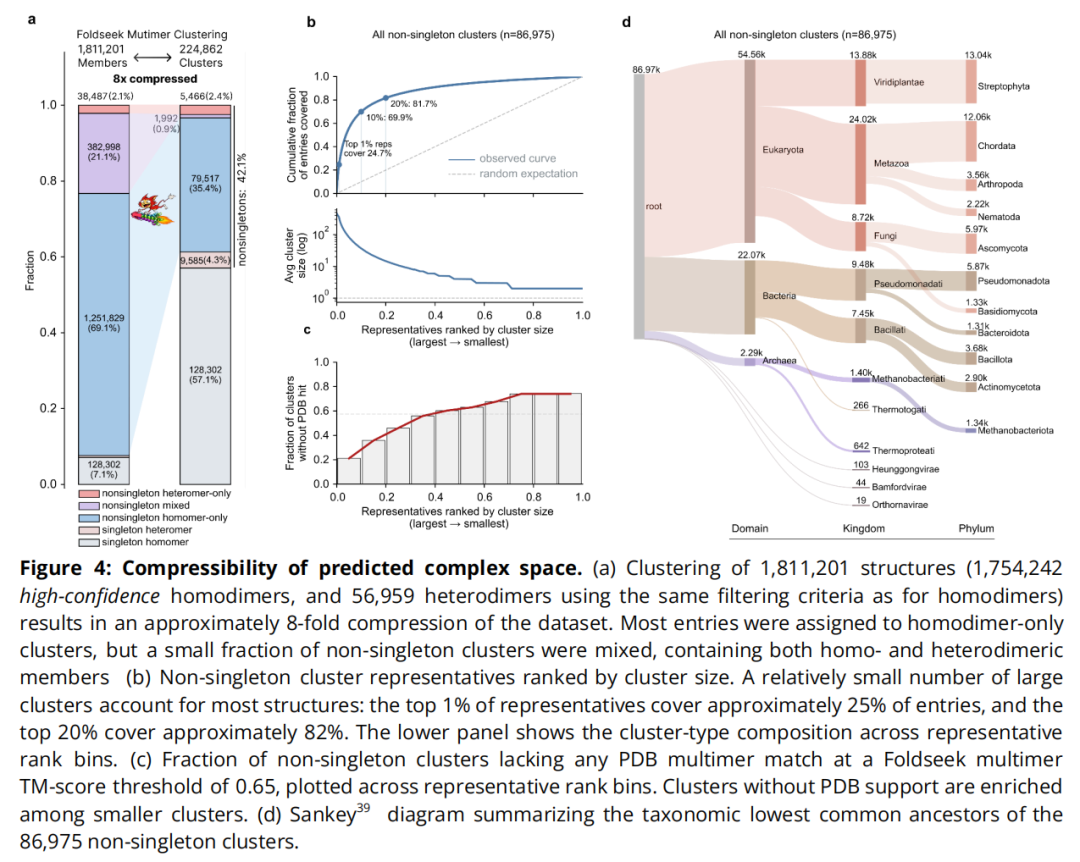
7.3 幂律分布特征
非单例聚类表现出显著的幂律分布:
- • 最大 1% 的聚类代表覆盖约 25% 的全部条目
- • 最大 20% 的聚类代表覆盖约 82% 的全部条目
这表明蛋白质复合物拓扑结构空间具有高度集中性——少数普遍性折叠模式主导了大量生物学实例。
7.4 与 PDB 已知复合物的结构重叠
以 Foldseek Multimer TM-score ≥ 0.65 为阈值搜索 PDB100:
- • 较大聚类与 PDB 实验结构重叠度高,结构上更普遍的解决方案倾向于与已知多聚体空间重合;
- • 较小聚类无 PDB 匹配的比例更高,代表当前 PDB 未覆盖的新颖拓扑结构,是后续实验验证的高优先级候选。
7.5 进化保守性分析
基于 NCBI Taxonomy 计算各聚类的最低共同祖先(LCA):
- • ~9% 的非单例聚类包含来自至少两个不同超界(细菌、古菌、真核生物)的成员,意味着这些复合物很可能起源于所有生命形式的共同祖先,代表跨生命域保守的基本功能模块。
八、代表性案例研究:复合物语境重新定义蛋白质结构
本节选取五个案例,展示四级结构预测相对三级结构预测的独特信息增益。
案例 1:折叠只在寡聚体语境中存在(结构域互换)
蛋白质: 盘基网柄菌(Dictyostelium discoideum)转录延伸因子 Eaf N端结构域蛋白(Q55DI5)
单体预测(AF-Q55DI5-F1) | 同源二聚体预测 | |
|---|---|---|
pLDDTavg | 50.56 | 86.06 |
结构特征 | β-折叠片段破碎,无完整折叠 | 通过结构域互换(domain swapping)形成完整折叠 |
两条链相互提供对方所需的结构元件,折叠横跨两条链而非存在于单条链内。Foldseek 搜索 PDB 发现相似架构(7okx),其折叠同样跨链形成,支持该预测的可靠性。
意义: 单体预测不仅低估,甚至可能给出根本错误的结构图像;结构域互换类折叠必须在复合物语境中才能被正确识别。
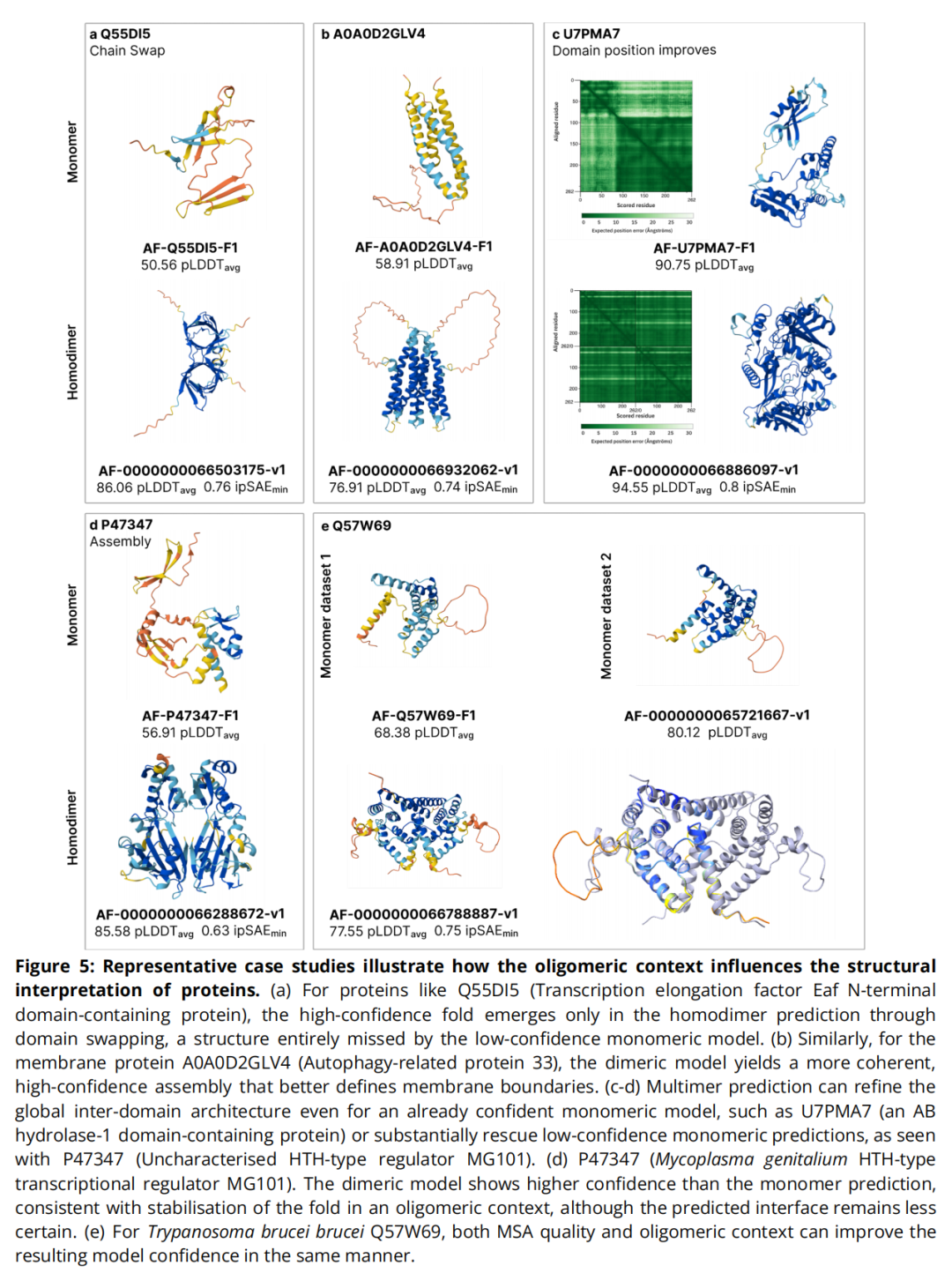
案例 2:膜蛋白寡聚化明确跨膜组装(膜蛋白拓扑)
蛋白质:Fonsecaea pedrosoi 自噬相关蛋白 33(A0A0D2GLV4)
单体预测 | 同源二聚体预测 | |
|---|---|---|
pLDDTavg | 58.91 | 76.91 |
ipSAEmin | — | 0.74 |
结构特征 | 四螺旋束存在但置信度低 | 两个四螺旋束形成连贯的跨膜组件,膜边界清晰 |
二聚体预测中,透射螺旋区域的空间跨度为 34–39 Å,与脂质双分子层核心厚度(~30–40 Å)高度吻合,合理界定了膜蛋白的跨膜范围。
意义: 对某些膜蛋白,单体预测可以恢复核心拓扑,但只有寡聚体模型才能充分解析完整组装体及其膜定位方式。
案例 3:优化结构域间相对排列(高置信度单体的进一步改善)
蛋白质:Sporothrix schenckii AB 水解酶-1 结构域蛋白(U7PMA7)
单体预测(AF-U7PMA7-F1) | 同源二聚体预测 | |
|---|---|---|
pLDDTavg | 90.75 | 94.55 |
ipSAEmin | — | 0.80 |
改善之处 | PAE 图中结构域间不确定性偏高 | PAE 矩阵不确定性显著降低,结构域间相对位置更精确 |
意义: 即使单体预测已有较高置信度,二聚体建模仍可通过约束结构域间相对排列提供额外信息,这对理解跨结构域的变构效应和界面细节尤为重要。
案例 4:二聚体预测"拯救"低置信度单体(折叠稳定化)
蛋白质: 支原体(Mycoplasma genitalium)HTH 型转录调控因子 MG101(P47347)
单体预测 | 同源二聚体预测 | |
|---|---|---|
pLDDTavg | 56.91 | 85.58 |
ipSAEmin | — | 0.63 |
HTH(Helix-Turn-Helix)转录因子普遍以二聚体形式与 DNA 结合,单体状态下折叠不完整符合预期。ipSAEmin = 0.63 偏低,作者认为这可能源于完整功能复合物还需要 DNA 参与,使界面预测存在固有不确定性。
意义: 对于生物学上以多聚体形式存在的蛋白质,强制以单体预测会导致低置信度,复合物预测可以从根本上改善结构质量并揭示其天然装配状态。
案例 5:MSA 质量与寡聚体语境的双重优化(被忽视物种)
蛋白质: 布氏锥虫(Trypanosoma brucei brucei)未表征蛋白(Q57W69)
模型 | pLDDTavg | 说明 |
|---|---|---|
原始单体(AF-Q57W69-F1) | 68.38 | 默认 MSA |
优化单体(AF-0000000065721667-v1) | 80.12 | 使用锥虫目专用 MSA(Wheeler lab 策略) |
同源二聚体(AF-0000000066788887-v1) | 77.55 | ipSAEmin = 0.75 |
优化单体与二聚体之间高度一致:链 A 叠合 RMSD = 0.67 Å(166 个修剪原子对)。
意义: 对于进化上代表性不足的物种,MSA 的质量是结构预测的关键瓶颈;寡聚体语境与高质量 MSA 可以相互验证,为被忽视热带病病原体提供可靠的结构假说。
九、数据可及性与工具链
9.1 AFDB 数据访问
- • 网址:alphafold.ebi.ac.uk
- • 已上线: 1,754,242 个高置信度同源二聚体(ipSAEmin ≥ 0.6、pLDDTavg ≥ 70、clashbackbone ≤ 10)
- • 即将上线: 约 2800 万个低置信度预测结果(FTP 批量下载)
- • 格式: ModelCIF 标准 mmCIF + Binary CIF,含界面评分元数据
9.2 计算工具开源
工具 | 获取地址 | 说明 |
|---|---|---|
ColabFold 1.6.0 | github.com/sokrypton/ColabFold | 含高通量改进 |
OpenFold(TRT+cuEq 版本) | github.com/aqlaboratory/openfold | Apache 2.0 |
cuEquivariance | docs.nvidia.com/cuda/cuequivariance | NVIDIA |
TensorRT | docs.nvidia.com/deeplearning/tensorrt | NVIDIA |
十、主要局限性与未来方向
10.1 已知局限
- 1. 异源二聚体置信度校准不足: 当前阈值源自同源二聚体验证,对典型异源二聚体(链间差异大)存在系统性偏倚,56,956 条异源结果仅作"暂定"处理。
- 2. 预测精度未达实验水平: AlphaFold-Multimer 存在已知偏差(如对某些界面几何的系统性错误),预测不能替代实验结构,但足以作为假说生成工具。
- 3. 仅覆盖二聚体: 本研究仅预测二聚体(同源 + 异源),更高阶的寡聚体(三聚体、四聚体及以上)未覆盖。
- 4. MSA 策略的影响: 异源二聚体采用简单拼接而非配对 MSA,可能对部分界面预测存在不利影响,尽管作者比较表明在高置信度阈值下差异不显著。
- 5. 计算失败率: 约 5% 的预测因内存溢出、序列过长或含非标准氨基酸而失败,未进一步分析。
10.2 后续工作方向
- • 异源二聚体置信度专项校准及更大规模发布
- • 完整 ~31M 预测结果的批量下载开放
- • 高阶寡聚体预测的探索
- • 变异效应分析(界面 SNP 功能影响预测)
- • 与 AlphaFold 3 等更新模型的集成
十一、对该领域的深远影响
11.1 对结构生物学的贡献
本工作实现了以下范式级突破:将蛋白质结构数据库从三级结构(单体)扩展至四级结构(复合物),并以此前实验数据库 1–3 个数量级的规模提供结构覆盖。案例研究表明,复合物语境不仅可以提升置信度,更可以根本性地改变对蛋白质折叠、组装和功能的解读。
11.2 对 AI 蛋白质科学的贡献
1,811,201 个带有标准化置信度指标的复合物结构,为下一代蛋白质设计模型(如 Proteina、Boltz-2 等)、蛋白质-蛋白质对接方法、界面功能预测模型提供了前所未有的大规模训练和基准数据集。
11.3 对全球健康的贡献
WHO 30 种优先疾病蛋白质组(含被忽视热带病病原体)的系统性结构覆盖,为结核、疟疾、锥虫病等疾病的药物靶点发现提供了结构基础,有助于缩小发展中国家疾病与发达国家疾病之间的研究资源鸿沟。
总结
维度 | 核心贡献 |
|---|---|
规模 | ~3100 万二聚体预测;180 万高置信度结构进入 AFDB |
物种覆盖 | 4,777 个蛋白质组,含 WHO 全球健康重点物种 |
方法论 | 系统性置信度校准(ipSAEmin);专用同源过滤 MSA 策略;GPU 加速后处理工具链 |
科学发现 | 结构域互换折叠仅在复合物中涌现;原核生物同源二聚化率显著高于真核;~9% 复合物跨超界保守 |
数据开放 | ModelCIF 格式、标准化元数据,支持搜索、可视化和批量下载 |
局限 | 异源二聚体校准待完善;高阶寡聚体尚未覆盖;预测精度不及实验 |
编者评价: 这是继 2021 年 AlphaFold2 论文和 AFDB 初始发布之后,计算结构生物学领域最具影响力的基础设施级工作之一。其意义不仅在于数量上的扩展,更在于将蛋白质结构研究的焦点系统性地推向了功能更为核心的"相互作用界面"层次,并为此提供了可公开访问、可重现、有标准化置信度注释的规模化数据资产。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-31,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号