Nat. Commun. | ClickGen:基于模块化反应与强化学习的可合成化学空间定向探索

Nat. Commun. | ClickGen:基于模块化反应与强化学习的可合成化学空间定向探索

DrugIntel
发布于 2026-04-13 17:50:37
发布于 2026-04-13 17:50:37

原文:ClickGen: Directed exploration of synthesizable chemical space via modular reactions and reinforcement learning 期刊:Nature Communications(2024)15:10127 DOI:https://doi.org/10.1038/s41467-024-54456-y 作者:Mingyang Wang†, Shuai Li†, Jike Wang†, Odin Zhang, Hongyan Du, Dejun Jiang, Zhenxing Wu, Yafeng Deng, Yu Kang, Peichen Pan, Dan Li, Xiaorui Wang, Xiaojun Yao, Tingjun Hou*, Chang-Yu Hsieh* 单位:浙江大学
目录
- 1. 研究背景与动机
- 2. 核心方法论
- 3. 模型技术细节
- 4. 基准测试结果
- 5. 物化性质与化学空间分析
- 6. PARP1靶点的湿实验验证
- 7. 讨论:创新、局限与展望
- 8. 方法学补充
- 9. 总结
一、研究背景与动机
1.1 AI驱动的从头药物设计(DNDD)现状
过去五年,基于深度学习(Deep Learning, DL)的从头分子生成方法迎来爆发式增长,仅报道的新方法就接近200种。代表性工作包括:
年份 | 研究团队 | 成果 | 关键时效 |
|---|---|---|---|
2019 | Aspuru-Guzik(GENTRL) | 设计DDR1激酶抑制剂 | 46天完成设计→合成→活性评测 |
2021 | IBM | 深度生成自编码器设计20个多肽生物分子 | 48天合成并验证 |
2021 | Schneider等 | LSTM结合微流控自动合成平台 | 25个分子中12个为LXR体外激动剂 |
2022 | Godinez等(VAE) | 疟疾PI4K靶点活性化合物 | 低纳摩尔水平活性 |
2022 | Li等(条件RNN) | 新型RIPK1选择性抑制剂 | 体外+体内双重验证 |
这些突破表明AI辅助药物设计正在从理论走向实验室实践。然而,一个核心瓶颈始终制约着该领域的工程转化:生成分子的合成可行性(synthesizability)普遍偏低。
1.2 核心问题剖析
当前分子生成模型主要分为两类范式:
配体为中心(Ligand-centric)方法
- • 代表:ChemVAE、REINVENT、GENTRL
- • 输入/输出:SMILES字符串或分子图
- • 引导信号:logP、合成可及性系数、QSAR活性预测值
- • 局限:忽略分子三维构象,与蛋白口袋的3D交互建模不充分
受体为中心(Receptor-centric)方法
- • 代表:RELATION、LIGAN、ResGen、SurfGen
- • 特点:显式建模蛋白-配体3D结构互作
- • 局限:合成可行性同样未充分保障
此外,已有若干**反应导向(reaction-based)**的生成模型尝试通过将合成子(synthon)按预定义反应规则组装来提升合成可行性,包括BBAR、SynNet和DeepLigBuilder+。但这类方法存在两大固有缺陷:
- 1. 实际合成困难:仅保证反应规则的形式合规,未考虑副反应、严苛反应条件、立体位阻等实际问题;原料稀缺或苛刻条件导致合成周期大幅延长
- 2. 生物活性不确定:以合成可及性为优化目标,但未充分保障所生成分子的真实生物活性,需依赖额外的FEP分析等间接手段评估
1.3 为什么选择点击化学(Click Chemistry)?
铜催化叠氮-炔烃环加成反应(CuAAC,Copper-catalyzed Azide-Alkyne Cycloaddition)是理想的合成导向生成规则,原因如下:
- • 反应条件温和:室温至60°C,铜(I)催化(CuBr、CuI或Cu(II)盐+抗坏血酸原位生成)
- • 溶剂兼容性强:水、乙醇、DMSO、THF等极性溶剂均可
- • 高选择性与高产率:通常在数分钟至数小时内完成,无需严苛纯化
- • 副反应极少:反应过程简洁,产物可预期
- • 工业可行性已验证:基于CuAAC规则的化合物数据库(如REAL数据库)已达数十亿量级,80%可合成率已获实验支持
结合另一模块化高效反应——酰胺化反应(DCC/EDC活化,温和极性溶剂条件,数分钟至数小时完成),ClickGen构建了兼顾多样性与可合成性的双反应框架。
二、核心方法论
2.1 ClickGen总体架构
ClickGen是一个三模块协同的合成导向分子生成系统:
┌──────────────────────────────────────────────────┐
│ ClickGen 系统 │
│ │
│ ┌─────────────────┐ ┌─────────────────────┐ │
│ │ 化学反应组合器 │ │ Inpainting生成模型 │ │
│ │ (CuAAC + 酰胺化) │ │ (U-net + BCT + LSTM)│ │
│ └────────┬────────┘ └──────────┬──────────┘ │
│ │ │ │
│ └──────────┬─────────────┘ │
│ ▼ │
│ ┌──────────────────────┐ │
│ │ 强化学习模块(MCTS) │ │
│ │ 奖励:Vina对接分数 │ │
│ └──────────────────────┘ │
└──────────────────────────────────────────────────┘核心设计哲学:通过inpainting技术解决合成可行性与新颖性之间的固有矛盾——反应组合器保障合成可行,inpainting模型赋予骨架跳跃能力,强化学习引导结合亲和力最优化。
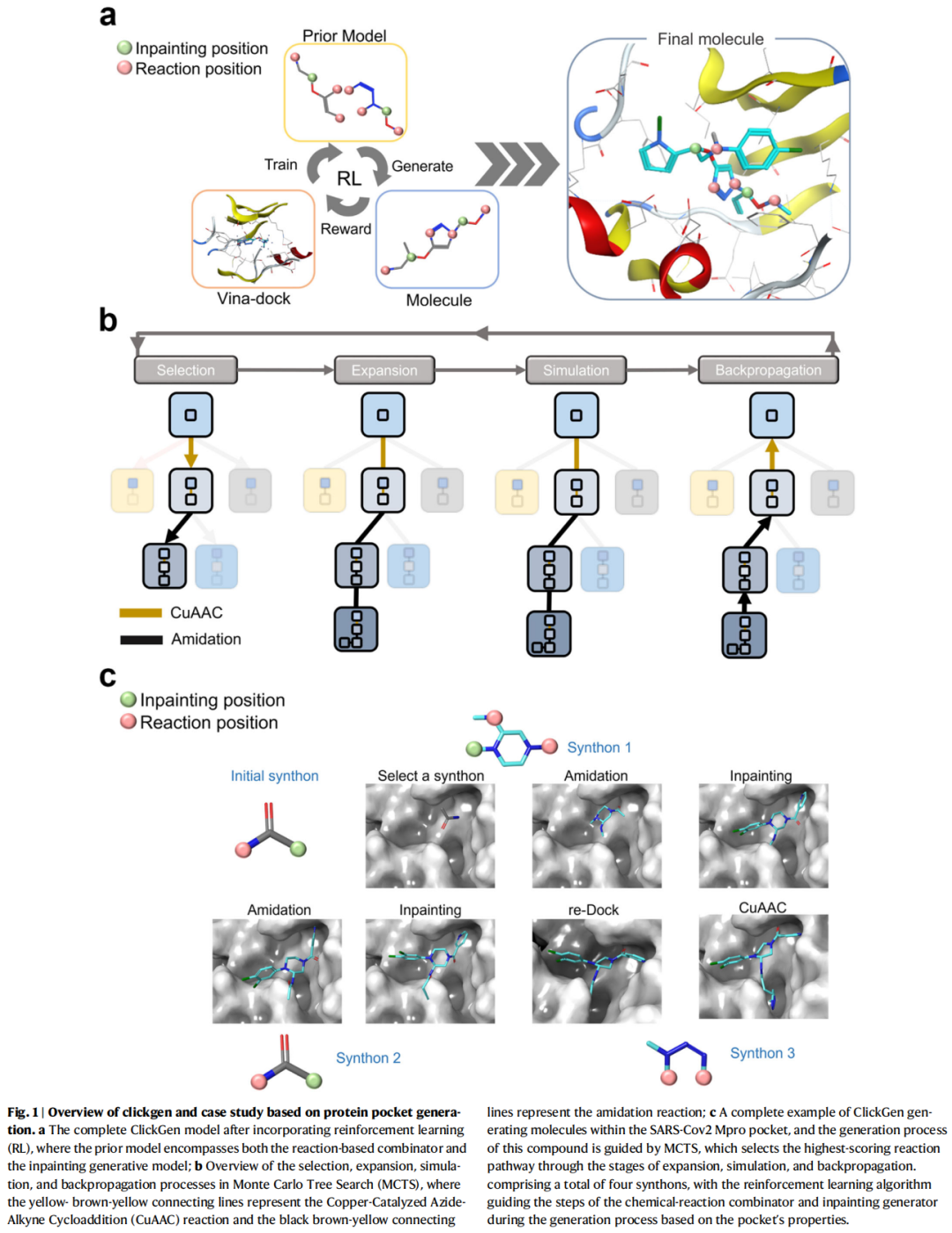
2.2 训练数据来源
- • 主训练集:Enamine REAL Diversity数据库(4820万化合物)
- • 过滤标准:−2 < logP < 7;MW < 500;HBA+HBD < 10;TPSA < 150;通过PAINS和MCFs过滤
- • 最终纳入:140万化合物用于模型训练
- • 靶点活性数据:BindingDB、ChEMBL、PDB
- • ROCK1抑制剂:917个(IC₅₀ 或 Kᵢ < 50 nM)
- • SARS-CoV-2 Mpro抑制剂:1203个
- • AA2AR拮抗剂:1154个
- • PARP1抑制剂:1667个
三、模型技术细节
3.1 化学反应组合器(Chemical Reaction Combiner)
功能:以负采样策略训练,使模型能够从合成子库中选择符合CuAAC和酰胺化规则的正确合成子进行拼装。
网络结构:
输入(合成子SMILES) → FC(128) → ReLU → FC(128) → Sigmoid → 输出概率p- • p ≈ 1:合成子选择正确(正样本)
- • p ≈ 0:合成子选择错误(负样本)
负采样策略(参考Word2Vec方法):
- • 正样本:与切分合成子Tanimoto相似度 > 0.7
- • 负样本:相似度 < 0.4
- • 最优正负样本比:1:10(100个正样本 + 1000个负样本)
关键发现:正负比低于10时,模型倾向于过度使用反应类型1和3,导致分子结构单调;比值超过10或同比扩大,则无法进一步提升。
损失函数:
3.2 Inpainting生成模型
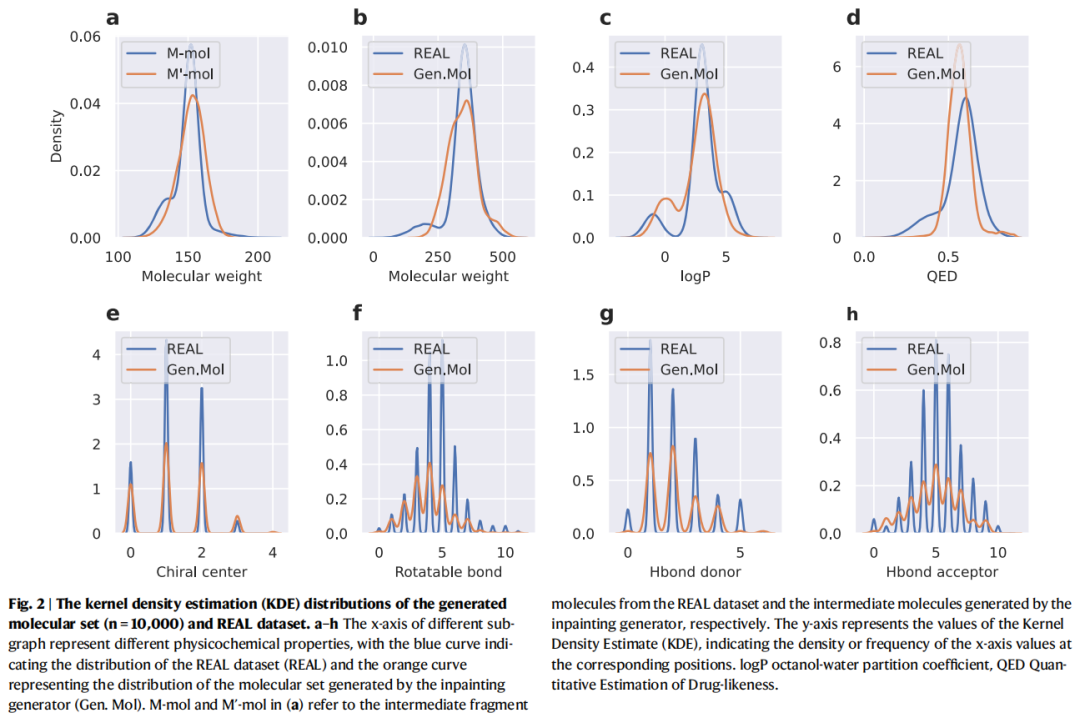
设计灵感:类比图像修复(Image Inpainting)——基于图像已知区域(边缘)推断被遮挡的中间区域,模型根据分子两端片段(、)生成合理的中间片段()。
网络结构:U-net启发的编码-解码架构,集成上下文注意力机制(Contextual Attention)
训练数据准备:
- • 从REAL数据库随机沿非环键切分分子为三段:、、
- • 使用Bemis-Murcko骨架对合成子进行遮罩(mask)
生成质量要求(通过损失函数约束):
- 1. 生成分子结构完整、化学合理
- 2. 物化性质与训练集分布一致
- 3. 保留反应化学键(酰胺键、三唑环)同时具备结构新颖性
- 4. 、与输入端保持一致
双向内容迁移(BCT):引入双向LSTM编码器()和解码器(),通过一致性损失约束中间片段与两端的平滑过渡:
总损失函数:
其中重建损失采用高斯权重掩码M(d),对距离边界较远的位置施加较小惩罚:
训练设置:Adam优化器,初始学习率1×10⁻⁴,训练50个epoch至收敛,约40小时(NVIDIA GeForce GTX 4090 GPU)
3.3 强化学习模块(Monte Carlo Tree Search, MCTS)
奖励函数:AutoDock Vina 4.2对接分数(越负越好)
MCTS四步流程:
- 1. 选择(Selection):按UCT公式选择合成子,兼顾exploitation与exploration 其中探索系数
- 2. 扩展(Expansion):若当前叶节点非终止节点,基于反应组合位点生成新的合成子节点并选择一个扩展
- 3. 模拟(Simulation):从扩展节点继续组装完整分子,获取最终奖励
- 4. 反向传播(Backpropagation):将奖励从扩展节点逐层传回至根节点,更新N和Q值:
两种先验模型对比:
- • ClickGen(无inpainting):仅使用反应组合器,依赖预定义合成子库
- • ClickGen-inpainting:组合器 + inpainting模型,支持生成新型合成子,实现骨架跳跃
四、基准测试结果
4.1 评估指标体系
指标 | 英文 | 含义 | 优化方向 |
|---|---|---|---|
有效性 | Validity | 生成SMILES有效率 | ↑ |
新颖性 | Novelty | 不在训练集中的比例 | ↑ |
唯一性 | Uniqueness | 去重后比例 | ↑ |
骨架数 | Scaffold count | 不同Bemis-Murcko骨架数量 | ↑ |
新颖骨架比 | Novel scaffold ratio | 新骨架占总骨架比例 | ↑ |
多样性 | Diversity | 1 − 平均Tanimoto系数 | ↑ |
FCD | Fréchet ChemNet Distance | 与训练集物化性质分布的距离 | ↓ |
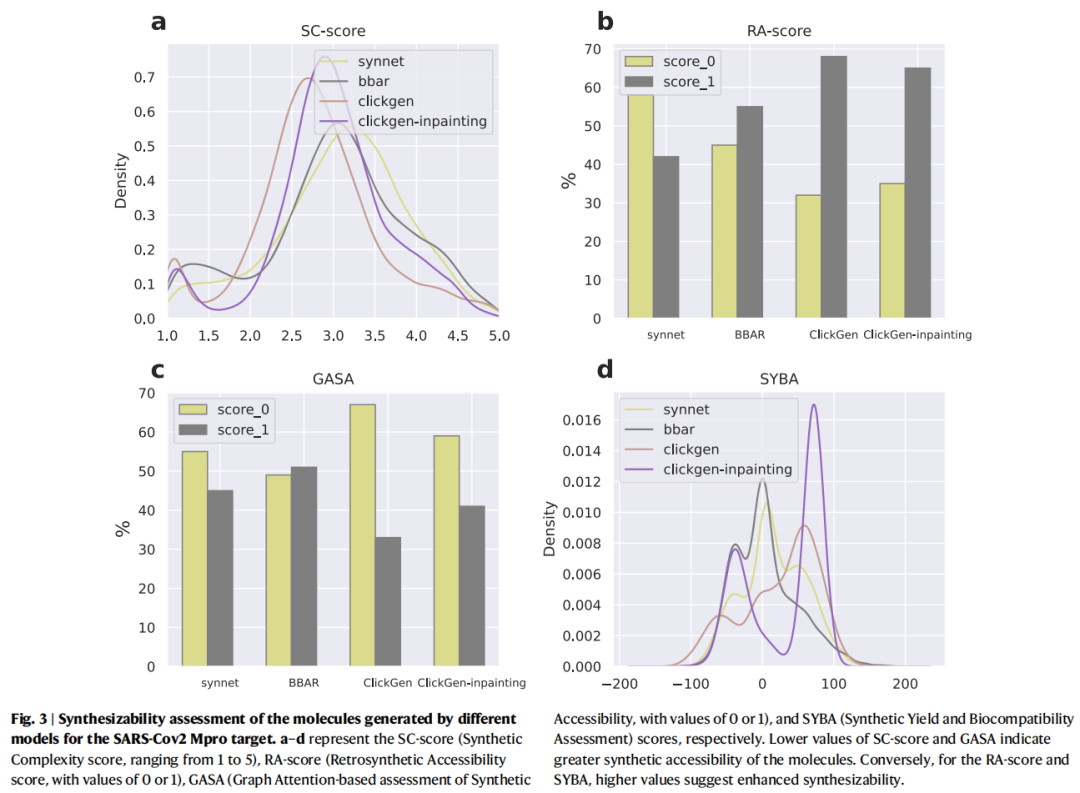
合成可及性评估采用四种互补指标:
指标 | 类型 | 范围 | 更优方向 |
|---|---|---|---|
SC-score | 描述符 | 1–5 | ↓(越低越易合成) |
RA-score | 逆合成 | 0–1 | ↑(越高越易合成) |
GASA | 图注意力 | 0–1 | ↓(0为易合成) |
SYBA | 贝叶斯 | 连续值 | ↑(越高越易合成) |
4.2 三靶点基准测试(n=10,000)
ROCK1靶点
模型 | 有效性 | 新颖性 | 唯一性 | 骨架数 | 新颖骨架比 | 多样性 | FCD↓ |
|---|---|---|---|---|---|---|---|
BBAR | 0.83 | 0.73 | 0.77 | 2450 | 0.18 | 0.53 | 17.42 |
SynNet | 1.00 | 0.86 | 0.52 | 2619 | 0.22 | 0.50 | 22.59 |
ClickGen | 1.00 | 0.71 | 1.00 | 2247 | 0.18 | 0.70 | 9.27 |
ClickGen-inpainting | 1.00 | 1.00 | 1.00 | 3758 | 0.53 | 0.77 | 9.06 |
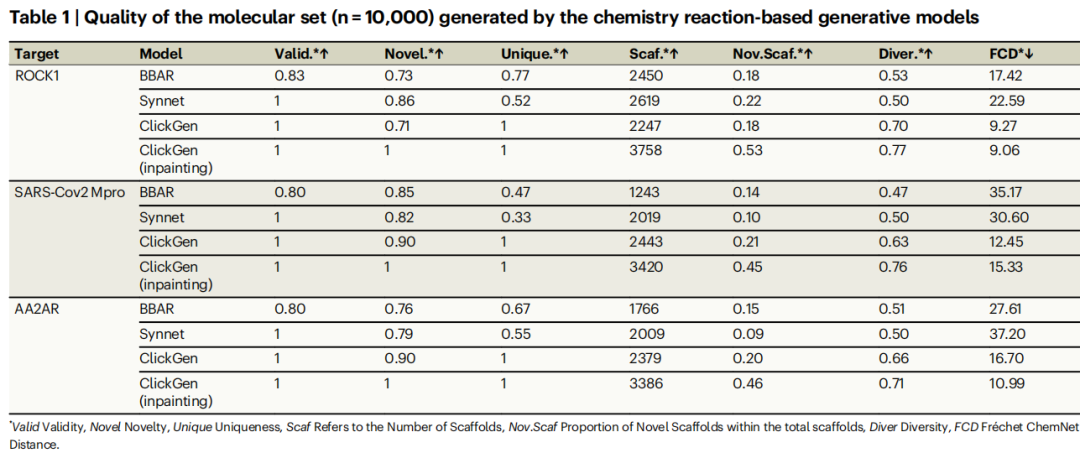
SARS-CoV-2 Mpro靶点
模型 | 有效性 | 新颖性 | 唯一性 | 骨架数 | 新颖骨架比 | 多样性 | FCD↓ |
|---|---|---|---|---|---|---|---|
BBAR | 0.80 | 0.85 | 0.47 | 1243 | 0.14 | 0.47 | 35.17 |
SynNet | 1.00 | 0.82 | 0.33 | 2019 | 0.10 | 0.50 | 30.60 |
ClickGen | 1.00 | 0.90 | 1.00 | 2443 | 0.21 | 0.63 | 12.45 |
ClickGen-inpainting | 1.00 | 1.00 | 1.00 | 3420 | 0.45 | 0.76 | 15.33 |
AA2AR靶点
模型 | 有效性 | 新颖性 | 唯一性 | 骨架数 | 新颖骨架比 | 多样性 | FCD↓ |
|---|---|---|---|---|---|---|---|
BBAR | 0.80 | 0.76 | 0.67 | 1766 | 0.15 | 0.51 | 27.61 |
SynNet | 1.00 | 0.79 | 0.55 | 2009 | 0.09 | 0.50 | 37.20 |
ClickGen | 1.00 | 0.90 | 1.00 | 2379 | 0.20 | 0.66 | 16.70 |
ClickGen-inpainting | 1.00 | 1.00 | 1.00 | 3386 | 0.46 | 0.71 | 10.99 |
关键结论:ClickGen-inpainting在所有靶点的新颖骨架比率约为SynNet的4–5倍,在保持低FCD(物化性质一致性)的同时实现骨架跳跃。
4.3 合成可及性评估(Mpro靶点,n=2000)
在四项合成可行性指标上,两种ClickGen模型均显著优于BBAR和SynNet。以最复杂的Mpro口袋为代表:
- • SC-score:ClickGen类模型评分更低(合成更简单)
- • RA-score:ClickGen类模型得分更高
- • GASA:ClickGen类模型接近0(高合成可及性)
- • SYBA:ClickGen类模型得分最高
重要发现:即便是采用inpainting产生新颖骨架的ClickGen-inpainting模型,其合成可行性指标也未出现明显下降,说明强化学习在筛选合成子时已隐性考量了化合物整体可合成性。
4.4 配体效率与构象分析
配体效率(LE):四种模型生成分子的LE均集中于0.2–0.3 kcal/mol/重原子,均处于高效配体设计区间,无明显"冗余原子"问题。
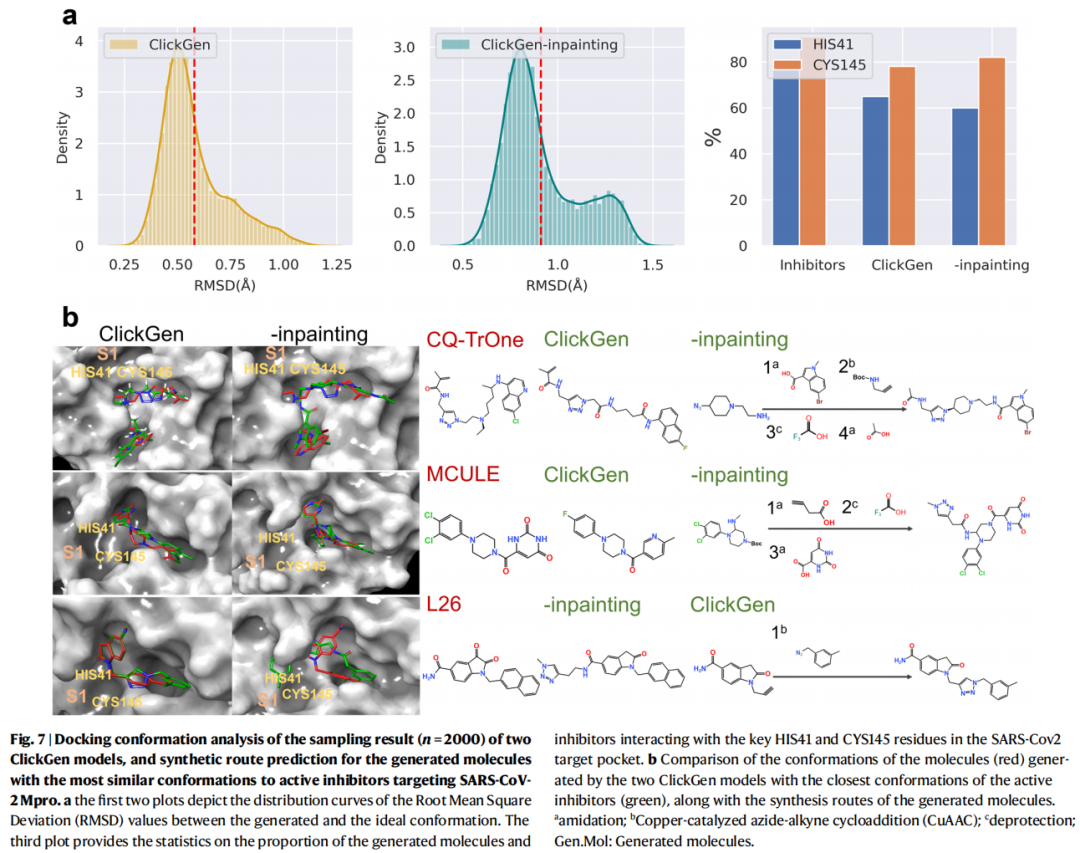
RMSD构象分析(Mpro靶点):
- • ClickGen(无inpainting):与理想结合构象的平均RMSD ≈ 0.6 Å
- • ClickGen-inpainting:平均RMSD ≈ 0.9 Å(构象多样性更大)
- • 两者均保持与S1区关键残基HIS41和CYS145的相互作用
关键残基交互分析:
残基 | 抑制剂参考 | ClickGen | ClickGen-inpainting |
|---|---|---|---|
HIS41 | 多类型 | 更多疏水作用 | 与抑制剂更接近 |
CYS145 | 多类型 | 与抑制剂匹配 | 氢键相互作用略少 |
GLU166 | 重要 | 两模型均复现 | 两模型均复现 |
GLN189 | 重要 | 略少 | 略少 |
特别说明:两种ClickGen模型均与SER46、GLU47、ASP48、MET49、LEU50形成更多相互作用,这类残基通常与微摩尔级活性化合物相互作用,提示部分生成分子可能仍需结构优化。
五、物化性质与化学空间分析
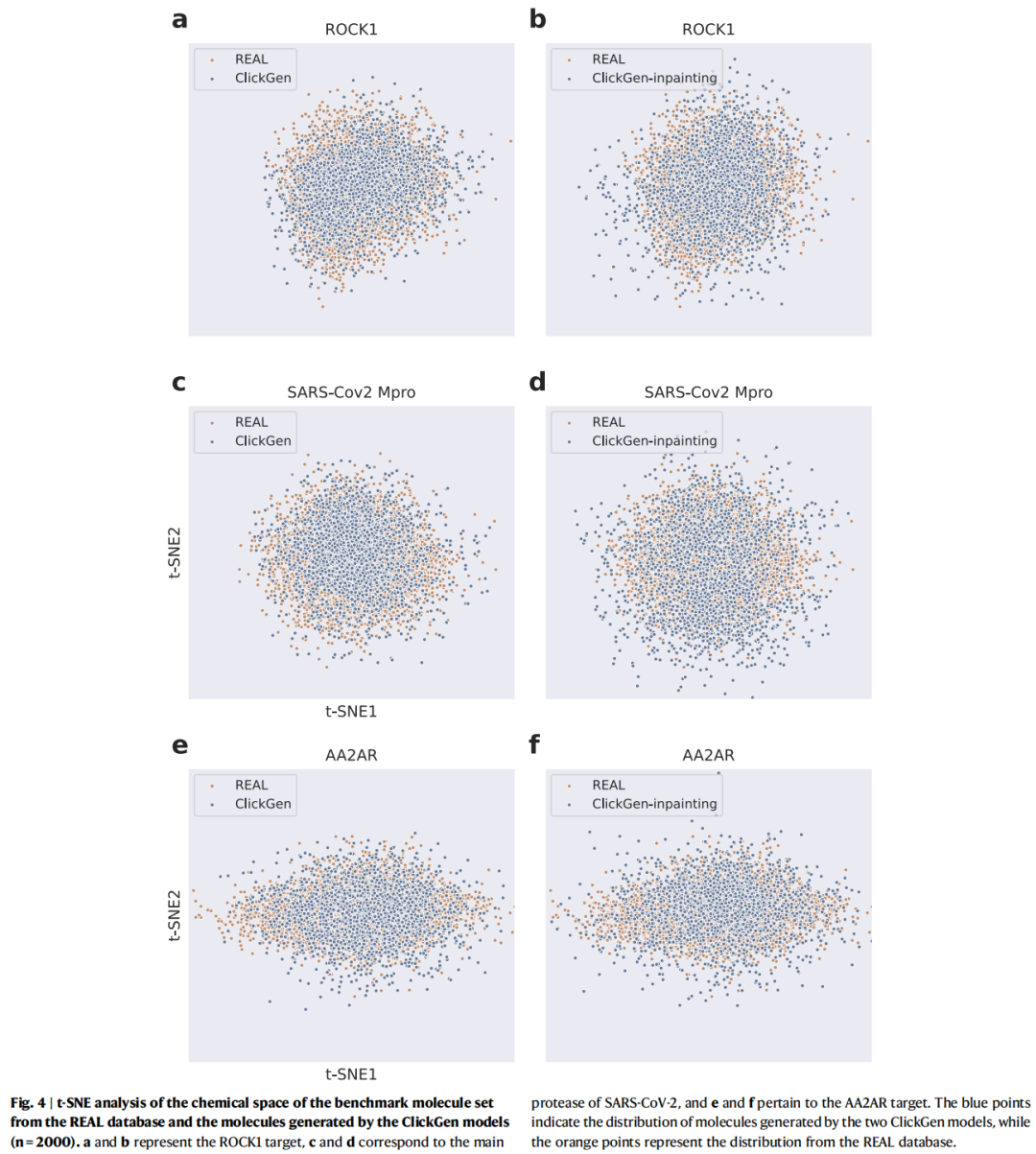
5.1 与REAL数据库的化学空间对比(t-SNE分析)
基于ECFP6指纹进行t-SNE降维分析,比较两种ClickGen模型生成的前2000个分子与REAL数据库虚拟筛选前10000个分子的化学空间分布:
- • ClickGen(无inpainting):已显著扩展至REAL数据库覆盖范围之外
- • ClickGen-inpainting:化学空间探索范围进一步扩大,与现有专利化合物重叠更少
这一特性对专利规避和全新骨架发现具有重要实用价值。
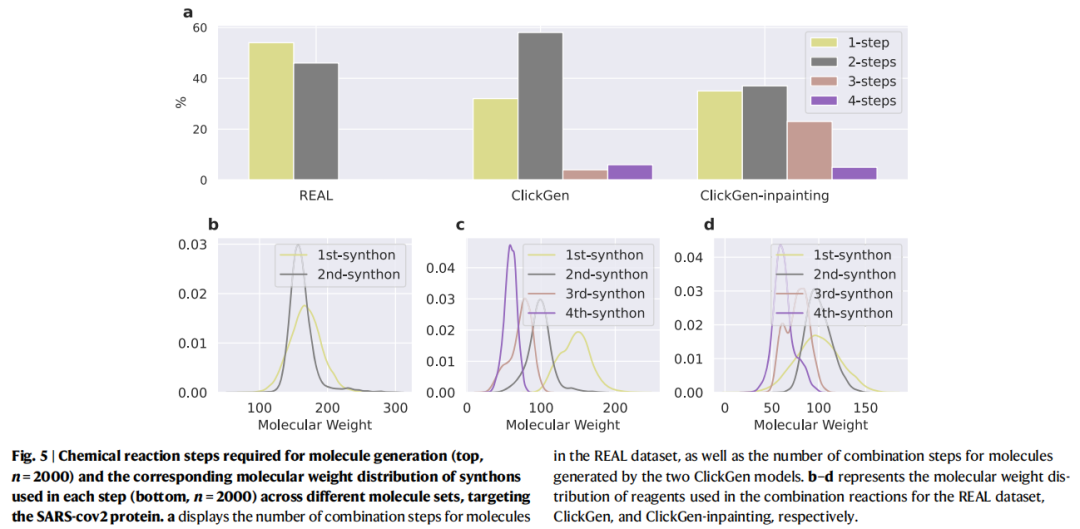
5.2 合成步骤分析
数据集 | 主要合成步骤数 | 含义 |
|---|---|---|
REAL基准集 | 1–2步 | 标准组合化学 |
ClickGen生成分子 | 3–4步 | 更复杂但仍可行 |
ClickGen-inpainting生成分子 | 3–4步 | 骨架更新颖 |
合成子分子量随步骤数增加而降低——第3、4步通常为末端化学基团的修饰,结合蛋白口袋形状进行灵活调整,但并不显著增加合成难度。
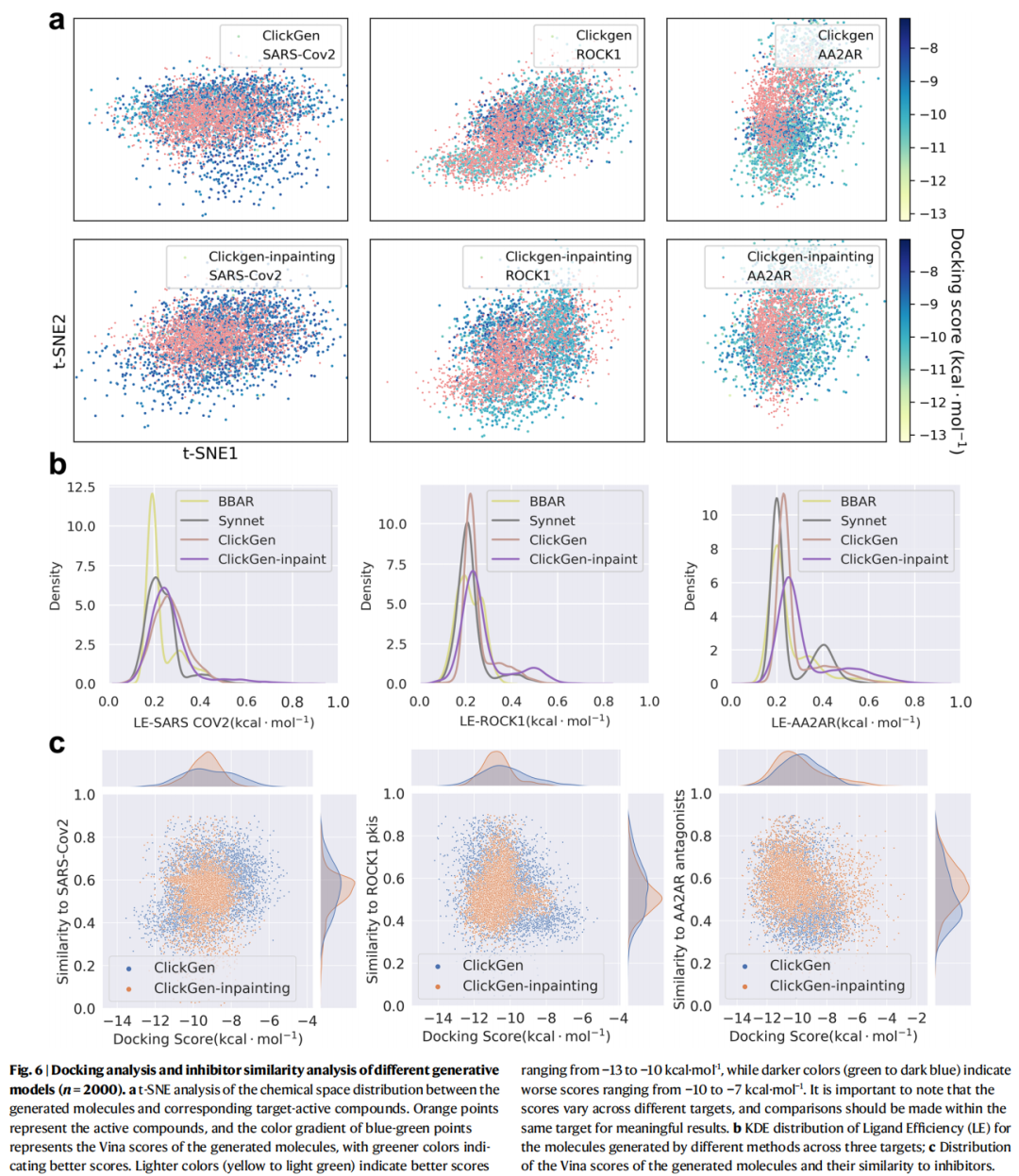
5.3 对接性能与相似性分析
对接分数均值对比:
- • ClickGen类模型(ROCK1):≈ −10 kcal/mol
- • ClickGen类模型(Mpro):≈ −9 kcal/mol
- • BBAR/SynNet:≈ −6 kcal/mol(显著逊色)
Tanimoto相似性分析:高对接分数分子通常与已知抑制剂具有高Tanimoto相似性,表明模型学习到了真实的活性药效团特征,而非随机得到低能量构象。
六、PARP1靶点的湿实验验证
6.1 研究背景
PARP1(Poly ADP-ribose Polymerase 1)是DNA损伤修复机制的关键酶,是重要的抗癌靶点,已有olaparib、rucaparib等多款抑制剂获FDA批准上市。本研究基于PARP1晶体结构(PDB ID:4BJC)对ClickGen-inpainting进行重训练,生成100,000个候选分子。
6.2 多级虚拟筛选流程
100,000个生成分子
↓ 新颖性/有效性/去冗余/骨架相似性过滤(Tanimoto<0.9)
~70,000个分子
↓ 物化性质筛选(250<MW<600;0<logP<8)
↓ 药效团匹配(Schrödinger):需与GLY863和SER904形成氢键
↓ Glide XP对接(前1%)
~700个候选化合物
↓ 骨架聚类 + 综合评估(相似性/合成可及性/知识产权潜力)
~30个化合物(跨低/中/高合成难度)
↓ 合成路线分析(原料可及性 + 反应条件评估)
3个先导化合物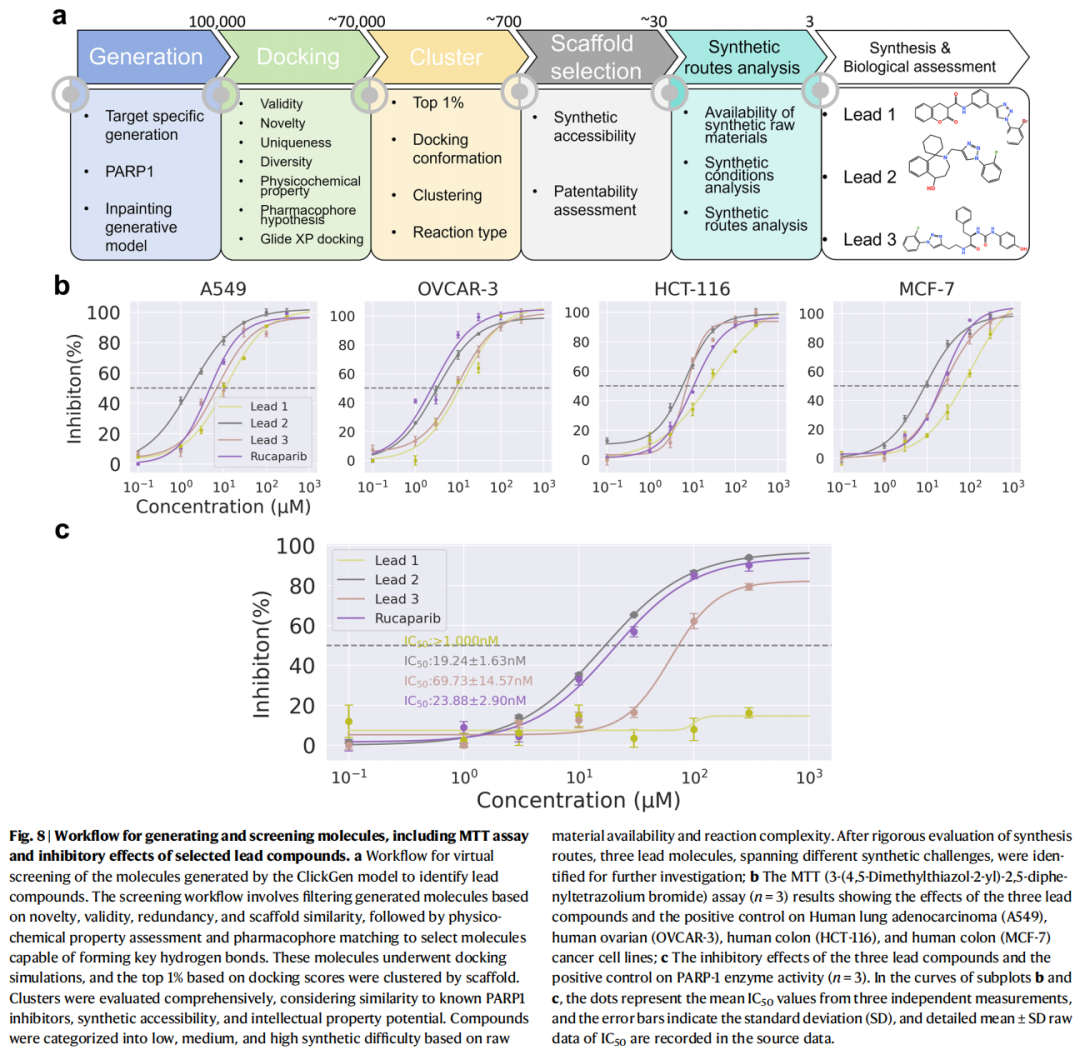
6.3 先导化合物特征
先导化合物1(Lead 1)
- • 合成难度:低(两步合成)
- • 反应类型:酰胺化 + CuAAC点击化学
- • 起始原料:香豆素-3-羧酸、3-乙炔基苯胺、1-叠氮基-2-溴苯(均商业可购)
- • 表征:Mp 201.4–201.8°C;ESI-HRMS [M+H]⁺ 490.3290(理论值),实测490.3342
先导化合物2(Lead 2)
- • 合成难度:较高(含需多步合成的复杂合成子)
- • 亮点:PARP1抑制活性优于阳性对照Rucaparib
- • 表征:Mp 166.6–166.9°C;ESI-HRMS [M+H]⁺ 407.2149(理论值),实测407.2254
先导化合物3(Lead 3)
- • 合成难度:中等(新型尿素键 + 多步组合 + 保护基策略)
- • 表征:Mp 244.1–244.7°C;ESI-HRMS [M+H]⁺ 489.1972(理论值),实测489.1797
6.4 生物活性评测
抗增殖活性(MTT法,四种癌细胞系)
细胞系涵盖:A549(人肺腺癌)、OVCAR-3(人卵巢癌)、HCT-116(人结肠癌)、MCF-7(人乳腺癌)
化合物 | A549 | OVCAR-3 | HCT-116 | MCF-7 | 毒性 |
|---|---|---|---|---|---|
Lead 1 | 低活性 | — | — | — | 偏高 |
Lead 2 | 强效 | 活性 | 强效 | 强效 | 较低 ✓ |
Lead 3 | 强效 | 活性 | 强效 | 强效 | 偏高 |
Rucaparib(阳性对照) | 参考 | 参考 | 参考 | 参考 | — |
PARP1酶抑制活性(体外,BPS Bioscience比色法试剂盒)
化合物 | IC₅₀ | 对比阳性对照 |
|---|---|---|
Lead 1 | > 1000 nM | 弱 |
Lead 2 | 19.24 ± 1.63 nM | 优于Rucaparib ✓ |
Lead 3 | 23.88 ± 2.90 nM | 接近Rucaparib |
Rucaparib | ~1000 nM(SI引用) | 参考 |
关键亮点:从ClickGen生成分子库→完成三个先导化合物合成并获得体外生物活性数据,全程仅用20天,远快于传统处理高新颖性分子所需的时间框架。
6.5 结构验证
所有先导化合物通过以下方法确认结构:
- • ¹H-NMR(600 MHz,DMSO-d₆)
- • ¹³C-NMR(150 MHz,DMSO-d₆)
- • ESI高分辨质谱(HRMS,Waters Q-TOF Premier)
七、讨论:创新、局限与展望
7.1 方法论创新总结
创新点 | 具体内容 |
|---|---|
反应规则选择 | 选择CuAAC+酰胺化两种模块化、高重复性反应,兼顾多样性与工程可行性 |
Inpainting引入 | 将图像修复思路迁移至分子生成,解决骨架新颖性与合成可行性的二元矛盾 |
闭环设计 | 强化学习以实验可测的对接分数为奖励,支持与自动化合成平台整合 |
合成路线随附 | 生成分子时同步给出可操作的合成路线,降低实验化学家的实施门槛 |
完整实验验证 | 罕见地完成从虚拟筛选→合成→体外活性评测的完整闭环,Lead 2达纳摩尔级活性 |
7.2 局限性与未来改进方向
- 1. 反应类型覆盖有限:目前仅覆盖CuAAC和酰胺化两类反应,部分生成骨架可能仍在现有专利保护范围内。计划引入更多易重复的模块化反应类型。
- 2. 依赖初始合成子库:无论是否使用inpainting,模型对公开合成子数据库(如REAL)存在较强依赖。计划增强inpainting的生成能力,减少对训练数据集的依赖。
- 3. 对接评分的局限性:以Vina对接分数作为活性代理存在内在偏差,不能完全反映真实结合自由能,部分高评分分子生物活性较弱(如Lead 1)。
- 4. 自动化合成集成:计划将ClickGen与自动化合成技术深度集成,通过新实验数据的持续反馈实现真正的闭环分子优化。
一句话总结
ClickGen通过将点击化学模块化反应规则、U-net启发的Inpainting分子生成和MCTS强化学习三位一体融合,在反应导向分子生成领域实现了新颖性与合成可行性的最优平衡,并以20天的完整实验闭环验证了其在真实药物发现场景中的可行性。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-09,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号