片段药物发现(FBDD)片段库设计原则:工业界20年经验的系统性总结

片段药物发现(FBDD)片段库设计原则:工业界20年经验的系统性总结

DrugIntel
发布于 2026-04-13 18:02:41
发布于 2026-04-13 18:02:41

文献来源: Keserű GM, Erlanson DA, Ferenczy GG, Hann MM, Murray CW, Pickett SD. Design Principles for Fragment Libraries: Maximizing the Value of Learnings from Pharma Fragment-Based Drug Discovery (FBDD) Programs for Use in Academia.J. Med. Chem. 2016, 59, 8189–8206. 作者机构: 匈牙利科学院自然科学研究中心、Carmot Therapeutics(美国)、GlaxoSmithKline(英国)、Astex Pharmaceuticals(英国)
导读
本文是一篇由来自多家顶级制药企业的科学家联合撰写的综述,旨在将工业界在片段药物发现(Fragment-Based Drug Discovery, FBDD)领域积累的系统性经验输出给学术界。作者们坦率地指出:FBDD在工业界已高度成熟,但在学术界往往执行不够规范,缺乏对最新设计原则的掌握,这正是本文写作的核心动机。
文章覆盖面极广,涵盖筛选技术选择与比较、片段库理化性质设计、分子复杂度理论、大小与形状考量、靶标相关策略、合成可优化性,以及库的物理管理等方方面面。
一、FBDD的核心逻辑:为什么是片段?
1.1 化学空间覆盖的数学优势
FBDD的根本优势来自组合数学的基本原理。一个包含 500,000 个化合物的高通量筛选(HTS)库,其化学空间覆盖率远不及一个仅含 数千个 精心选择片段的FBDD库。这是因为:
- • 分子量 ≤ 250 Da 的有机小分子,其理论可能数量约为 10⁷ 量级(可实际合成的化学空间有限);
- • 而药物样分子(MW ~500 Da)的化学空间估计高达 10⁶⁰ 量级,HTS根本无法有效采样;
- • 片段通过低分子量、低复杂度的特性,指数级提升了对化学空间的覆盖效率。
这意味着:几乎可以确信,片段库中存在能与任何蛋白质口袋发生有效相互作用的分子。
1.2 "弱结合"的战略意义
片段通常对靶蛋白的亲和力极弱(Kd在100 μM至10 mM范围),这在传统HTS思维中是"失败"——但在FBDD中,这恰恰是设计特性而非缺陷:
- • 苗头片段往往展现出高质量的焓驱动(enthalpy-driven)结合,与靶蛋白形成精准、定向的极性相互作用(尤其是氢键);
- • 由于结构小、与蛋白的接触面有限,片段结合时几乎不存在"无效"的空间冲突;
- • 片段可通过基于结构的药物设计(SBDD)逐步生长为高亲和力先导物,整个过程中配体效率(Ligand Efficiency, LE)更易维持;
- • 片段的命中率(hit rate)本身也是评估靶标"可成药性(ligandability)"的定量指标。
1.3 FBDD与HTS的本质区别
维度 | HTS | FBDD |
|---|---|---|
化合物数量 | 10⁵–10⁶ | 500–3000 |
分子量范围 | ~400–500 Da | ~140–230 Da |
典型亲和力 | <10 μM | 100 μM – 10 mM |
命中检测方法 | 生化活性为主 | 生物物理方法为主 |
化学空间覆盖 | 窄 | 宽 |
先导物质量 | 参差不齐 | 通常LE更高 |
优化路径 | 基于活性SAR | 基于结构的SBDD |
二、片段库设计的总体框架
2003年,Congreve等人从有限的片段筛选数据中归纳出著名的"三规则"(Rule of Three, RO3):
分子量 ≤ 300 Da,氢键供体数 ≤ 3,氢键受体数 ≤ 3,cLogP ≤ 3
然而,文章作者们明确指出:RO3是起点,而绝非全部。十余年实践表明,成功的片段库设计需要在以下多个维度上综合考量。
片段库设计的四项核心原则
- 1. 化学空间采样:库中片段须覆盖关键的结合药效团(pharmacophore),以确保能与蛋白质热点(hot spot)产生有效互动;
- 2. 大小与复杂度的平衡:片段过大则命中率下降(复杂度过高),过小则结合模式不稳定(无法建立可靠SAR);
- 3. 合成可扩展性:每个苗头片段需有充足的生长向量(growth vector),使后续优化具备可行的合成路径;
- 4. 质量控制:系统排除PAINS(泛筛活性干扰化合物)、聚集体形成化合物、高反应性官能团,并严格管控纯度与溶解度。
三、筛选技术的选择与比较
3.1 为什么生化筛选不够用?
HTS常用的生化活性检测通常只能检测 Kd < 10 μM 的结合,而片段的典型亲和力比这弱100–1000倍。因此FBDD必须依赖更灵敏的生物物理学技术。
3.2 六大主流技术的系统比较
技术 | 灵敏度下限 | 特异性评估 | 通量 | 结构信息 | 假阳性/假阴性倾向 |
|---|---|---|---|---|---|
表面等离子共振(SPR) | 高 μM | ✓ | 中 | 无 | 中假阳性 |
配体观测NMR(STD-NMR等) | 低 mM | ✗ | 中 | 少 | 中假阳性 |
蛋白观测NMR | 低 mM | ✓ | 低 | 高 | 低假阳/假阴 |
X射线晶体学 | 中 mM | ✓ | 低 | 极高 | 低假阳/高假阴 |
热位移(TSA/DSF) | 高μM–低mM | ✗ | 高 | 无 | 高假阳/假阴 |
生化活性检测 | 高 μM | ✓ | 高 | 无 | 高假阳/假阴 |
3.3 技术特性的深层解读
SPR 能检测结合动力学,但芯片表面的非特异性结合是主要假阳性来源;配体构型变化也可能影响信号。
配体NMR(STD-NMR, WaterLOGSY) 对弱结合非常灵敏,适合混合物(cocktail)筛选,但无法区分特异性与非特异性结合,需正交验证。
蛋白NMR 是特异性最高的技术之一,可直接定位结合位点,但对蛋白用量要求高(需同位素标记),通量低。
X射线晶体学 提供原子分辨率的结合模式,假阳性极低,但假阴性率高(需要化合物有足够的晶体占有率)。随着自动化晶体学平台的发展(如CSIRO的协作结晶中心),其通量正在显著提升。
热位移(TSA) 操作简便、通量高,但对化合物降低Tm的情况难以解读,特异性较差。
生化活性检测 在排除聚集干扰、设置正确浓度的前提下可用于FBDD,但需更严格的后续验证。
3.4 多靶标案例中的关键发现
文章通过六个具体案例(HIV整合酶、Checkpoint激酶2、p38α激酶、MMP12/胰蛋白酶、HSP90、内囊蛋白酶)揭示了一个核心结论:
不同技术筛选出的苗头化合物重叠度极低。
典型数据:
- • HIV整合酶筛选中,500个片段经STD-NMR识别62个命中,经SPR识别16个命中,两者无重叠;
- • Checkpoint激酶2筛选中,生化法(1.1%命中率)与热位移法(3.4%命中率)发现12个共同命中,但各自有31和49个独有命中;
- • HSP90筛选中,NMR与SPR命中重叠度达90%,但与热位移法的重叠度仅74%。
实践结论:
- • 正交筛选(≥2种技术)是必要的;2011年平均使用2.4种技术,到2013年已提升至3.6种;
- • 生化法+生物物理法的组合优于两种生物物理法的组合(Novartis大规模元分析,35个筛选活动);
- • 不应要求苗头物在所有技术中均显示活性,否则等于用最不灵敏的方法决定最终筛选下限。
3.5 技术选择对库设计的反向约束
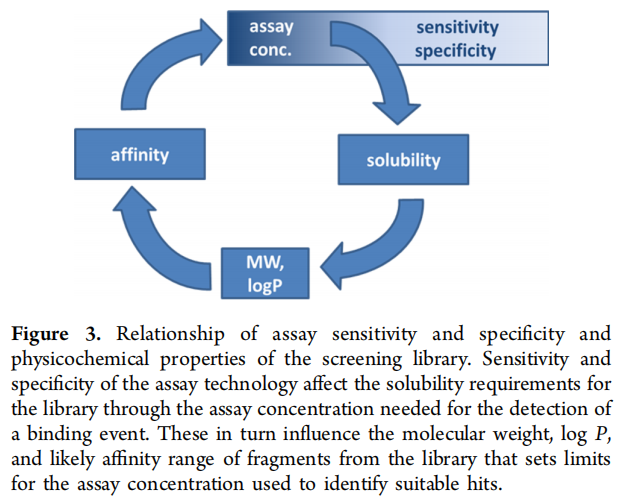
筛选技术的选择反向决定了片段库的物化性质要求(图3所示循环关系):
assay sensitivity & specificity
↓
assay concentration requirement
↓
solubility requirement
↓
MW & logP constraints
↓
accessible affinity range例如:NMR和X射线晶体学通常在 mM 级浓度下筛选,因此对溶解度要求极高;而SPR在100 μM–500 μM级别操作,相对宽松。
四、分子复杂度理论与配体效率
4.1 Hann复杂度模型
Hann等人于2001年提出的分子复杂度模型是FBDD的理论基石之一。该模型用两条概率曲线描述片段结合行为:
- • 配体匹配概率(蓝线):随复杂度增加而单调下降——分子越复杂,与靶蛋白"完美匹配"的概率越低;
- • 可检测结合概率(红线):随复杂度增加而先升后降——太小无法产生足以检测的结合能;
- • 有效事件概率(绿线)= 两者之积,在中等复杂度(约6–8个相互作用位点)时达到峰值。
关键推论:
- • 水分子是极端案例——作为55.5 M的"超高浓度片段",以最多四个定向氢键与蛋白精确结合,是复杂度最低、匹配概率最高的"完美片段";
- • 片段的4–6个非氢原子相互作用点,恰好处于"有效事件"概率最高的区间;
- • Astex数据显示:多个筛选活动中,命中片段的HAC众数为12,而实际筛选库的HAC众数为14——说明更小的片段命中率更高。
4.2 复杂度与信息内容
从信息论视角,不同类型的分子相互作用具有不同的"信息内容":
- • 高信息内容:定向极性相互作用(氢键、盐桥)——空间精确性高,难以"滑入"错误位置;
- • 低信息内容:亲脂相互作用——无方向性,可与蛋白表面多处非特异性结合;
这一分析解释了为何:
- 1. 含强氢键锚点的片段在优化过程中更易保持结合模式;
- 2. 早期依赖亲脂性提升活性("亲脂性增强假象"),会导致后期优化中出现广泛的非特异性结合,是FBDD中最常见的陷阱之一;
- 3. 芳香环在片段结合中高度普遍——其固有极化率(polarizability)使其能适应不同的结合环境,兼具一定方向性和灵活性。
4.3 配体效率指标体系
文章推荐在FBDD不同阶段使用不同的效率指标:
配体效率(LE):
可接受下限:LE ≥ 0.3(对良好结合位点而言)。
亲脂性配体效率(LLE / LipE):
优先选择LLE高的苗头物,避免亲脂性驱动的虚假活性。
重原子校正的亲脂配体效率(LLEAT):
兼顾大小、亲脂性与活性,与LE量纲一致,推荐在先导物优化阶段使用。
基团效率(Group Efficiency, GE):
评估每个优化步骤新加原子的"效率",避免盲目增加分子量。
实践警示: 这些指标是指导性工具而非绝对法则。文章明确提醒:指标应作为过滤和优先级排序的辅助手段,不应僵化地作为go/no-go决策依据。
五、片段的大小与三维形状
5.1 最优尺寸范围的共识
文章梳理了关于片段最小有效尺寸的大量实验证据,形成如下共识:
- • 重原子数 10–20:这一范围内的片段能够特异性结合多种蛋白质口袋(包括极性口袋和蛋白-蛋白相互作用界面),并在生长为先导物的过程中较大概率保持结合模式;
- • Practical Fragments调查:超过85%的受访者认为最小片段尺寸应在5–10重原子之间;
- • 晶体学筛选经验:含10–14个重原子的片段命中率最高;
- • 极小片段(<10重原子)理论覆盖化学空间更广,但结合模式太过多变,难以建立有效SAR。
5.2 结合模式保守性——从理论到实证
片段优化的核心假设是:随着片段"生长",初始结合模式得以保持。但实验证据表明,这一假设并不总是成立:
支持保守性的证据:
- • pVHL:HIF-1α相互作用抑制剂拆解研究:最小13重原子片段仍保持原始结合姿态;
- • pp60src SH2结合域:11重原子的苯磷酸盐保持结合位点及氢键网络;
- • Orita等分析25个优化案例:片段与先导物核心结构的RMSD均低于1 Å,氢键模式完全保留。
挑战保守性的证据:
- • Bcl-XL抑制剂拆解:片段未保持原有相互作用,全部聚集在蛋白位点的单一区域;
- • β-内酰胺酶抑制剂拆解(8、11、14重原子片段):均未保持完整抑制剂中对应基团的结合模式;
- • Kozakov等提出解释:结合模式是否保守,取决于片段与蛋白"主热点"(primary hot spot)的重叠程度。
Kozakov热点理论的实践意义:
- • 使用计算溶剂映射(computational solvent mapping,如FTMap服务器)预判主热点位置;
- • 优先选择与主热点有良好重叠的片段进行优化,此类片段的结合模式在结构扩展中更稳定;
- • 次要结合位点的片段,结合模式在优化后更易发生漂移,需要贯穿优化全程的结构确认。
5.3 三维形状多样性——机遇与代价
主流做法的局限: 传统片段库以含平面芳香环(如嘧啶、吡啶)的化合物为主,库的三维形状多样性严重不足。
增加3D性的潜在收益:
- • 降低非特异性结合(减少"分子肥胖");
- • 提升溶解度;
- • 覆盖更多生物相关化学空间,对难靶标(如PPI界面)更有利。
增加3D性的实际代价:
- • 分子复杂度上升,命中率下降(Astex数据证实3D片段命中率低于平面化合物);
- • 立体中心的引入显著增加合成难度;
- • 需要更大的库来弥补单个片段命中率的降低。
文章的平衡建议: FBDD的最大化利用取决于复杂度、尺寸与多样性之间的精细平衡,应根据靶标特性和筛选技术灵活调整3D片段的比例,而非一概而论。
六、靶标相关策略
6.1 可成药性(Ligandability)评估
文章区分了两个常被混淆的概念:
- • Druggability(可成药性):靶标是否能被药物调控且具有治疗价值;
- • Ligandability(可配体性):靶标是否能被小分子结合,不论其最终是否成为药物靶标;
FBDD最有价值的用途之一,正是在靶标验证早期评估可配体性——片段筛选的命中率与HTS命中率及最终先导物优化成功率均具有良好相关性。
6.2 不同靶标类型的FBDD策略
酶类靶标(最有利):
- • 进化为结合小分子(底物/辅因子),活性位点口袋明确;
- • 以激酶为例,ATP结合位点对片段高度开放;但定向库(如激酶专项库)可能牺牲新颖性;
- • GSK Kinase库研究(1064个片段,30种激酶)揭示:即使是"应该无选择性"的腺嘌呤,也只对不到半数激酶有强抑制——片段选择性常被低估。
蛋白-蛋白相互作用(PPI)靶标(挑战最大):
- • 结合面大而平,缺乏深口袋;
- • 需要更高的筛选浓度(有时达到10 mM),配体效率通常较低;
- • 成功案例:AbbVie的BCL-xL工作(最终获批Venetoclax);Astex的cIAP1/XIAP双重抑制剂;
- • 策略建议:在更高浓度下筛选,接受低LE的初始苗头物,以结构为导向进行深度优化。
变构位点(被低估的机会):
- • 片段因其小尺寸,特别适合探测隐蔽的(cryptic)变构口袋;
- • Novartis PAK1变构抑制剂:相对441种激酶,选择性>50倍;
- • HIV逆转录酶晶体学筛选:一次性发现16个结合位点;
- • 理论优势:变构抑制剂天然具有更高选择性,且可调控HTS难以发现的调控蛋白。
膜蛋白靶标:
- • 传统FBDD困难重重;
- • 热稳定化突变(thermostabilized mutants) 策略的出现显著改善了情况,使膜蛋白可接受SPR、热位移和X射线晶体学筛选;
- • GPCR领域已有成功案例(mGlu5阴性变构调节剂HTL14242等)。
6.3 目标导向库 vs. 通用库的争论
目标导向库的优势: 命中率高,化学优化路径清晰(如激酶ATP竞争性片段库的成功)。
目标导向库的风险: 新颖性不足,可能错过最有价值的意外发现(serendipity)。典型反例:PAK1筛选发现的变构片段与腺嘌呤几乎无结构相似性,却产生了高度选择性抑制剂。
文章结论: 两种策略各有其用,关键是目的。若时间紧迫、靶标明确,用导向库;若探索新靶标或新机制,用多样性通用库。
七、合成相关考量
7.1 "可优化性"的系统设计
一个理想的FBDD片段不仅要能"命中"靶蛋白,更要"长得出来"——即具备充足的合成生长向量(synthetic handle),能通过2–4步合成扩展为先导物。
文章介绍了多种系统化确保可优化性的策略:
反应字典法(Reaction Dictionary): Novartis团队建立了反应词典,专门筛选那些关键活性基团被"遮蔽"(masked)、可通过简单去保护/转化暴露合成把手的片段。
Poised Fragment Library(预置片段库): Cox等人设计的片段库中,所有片段均直接来自简单合成反应,使类似物的快速制备成为可能;此方法已成功用于PHIP(2)溴结构域的首个抑制剂发现。
RECAP方法: 将片段拆解为可扩展的"核心"结构,通过子结构搜索评估已有取代位点的数量,从而在筛选前完成优化潜力排序。
可优化性的计算评估(In Silico Assessment): 基于内部及外部数据库的子结构搜索,对候选片段的优化可行性进行虚拟排序,适用于处理大量潜在片段。
7.2 特种片段库的设计思路
3D片段(多样性导向合成,DOS):
- • Hung等人通过DOS策略生成三维片段,覆盖主成分惯量(PMI)空间中传统库欠采样的区域;
- • 商业上可通过3D Fragment Consortium(>500个化合物)获取。
天然产物来源片段:
- • 天然产物天然富含sp³碳,立体中心多,三维性高;
- • Over等人系统分析天然产物,提取高sp³含量的片段核心,筛选发现了p38激酶的非芳香性变构片段;
- • 注意事项:部分天然产物结构含PAINS基团(如醌类、邻苯二酚),直接使用时需严格过滤。
共价片段库(Covalent Fragment Library):
- • 不可逆共价(Irreversible Covalent):Statsyuk课题组的丙烯酸酯片段库,已对多个蛋白实现选择性抑制;
- • 可逆共价(Reversible Covalent):Taunton课题组的氰基丙烯酰胺片段,可与半胱氨酸形成可逆共价键,已发展出高选择性激酶抑制剂,兼顾效力与安全性;
- • Tethering法:基于热力学驱动的二硫键交换,适用于在特定半胱氨酸附近精准筛选,Genentech用于K-Ras靶点探索。
八、片段库物理管理
这部分内容在综述中较为罕见,但作者专设章节介绍,足见其实际重要性。
8.1 质量控制:入库标准
纯度要求: 每个片段须达到90–95%以上纯度。最低标准为HPLC-MS确认,推荐同时使用NMR,尤其在NMR为筛选技术时(可将入库纯度确认与筛选前处理合并)。
商业来源的警示: 针对超过10家供应商的>10,000个样品调查显示,16%的样品未通过QC,部分供应商失败率高达33%。因此,即使采购商业片段库,也必须进行in-house重新确认。
备用干储(Dry Stock)的重要性: 需为后续确认实验保留足量固体原料。Pfizer标准要求每个自有化合物储备至少200 mg。商业来源片段须在采购时确认长期供货能力,否则可能出现苗头物确认时货源已断的困境。
8.2 储存溶剂的选择
DMSO(绝大多数情况下的首选):
- • 溶解度高,与绝大多数蛋白质筛选系统兼容;
- • 通常配制浓度50 mM以上(确保最终assay浓度为1 mM时,DMSO含量≤2%);
- • 若NMR为筛选手段,直接使用氘代DMSO(DMSO-d₆),额外成本极小但节省后续操作;
- • Monash大学团队采用200 mM高浓度储液,进一步降低有机溶剂引入量,但存在更多化合物沉淀风险。
甲醇(晶体学特定用途):
- • 挥发性使其适合直接加入晶体化板,不改变最终缓冲液成分;
- • 低表面张力导致精确分液困难;化合物干燥后溶解性变差;实际应用有限。
8.3 储存温度与稳定性管理
DMSO溶液的降解风险(基于Procter & Gamble对7200个化合物的研究):
储存时间 | 室温降解率 |
|---|---|
3个月 | ~8% |
6个月 | ~17% |
12个月 | ~48% |
冻融循环的隐患: 反复冻融会引入大气水分(DMSO极易吸湿),导致片段溶解度急剧下降,甚至发生沉淀。实验表明,经历反复冻融的样品比持续冷冻或室温保存的样品浓度更低。
推荐策略:
- • +4°C或-20°C低温保存(2014年Practical Fragments调查:超过50%的受访者采用此方案);
- • 惰性气体(氮气/氩气)封存,低湿度环境;
- • 每年至少进行一次库完整性重新评估;
- • 苗头物确认实验必须使用新鲜配制样品。
化合物本身的稳定性风险:
- • DMSO具有轻度氧化性,部分化合物在其中会快速降解;
- • 看似稳定的骨架(如苯并噁唑)在固体状态下仍可发生水解;
- • 某些化合物在DMSO中自发环化形成活性物种,而该活性物种在水溶液中又不稳定,使assay解读极为复杂。
九、片段库构建推荐参数总表
以下参数是本文最具实操价值的核心产出(基于表6,结合全文内容整合):
9.1 基础物化性质
参数 | 推荐范围 | 注释 |
|---|---|---|
分子量(MW) | ~140–230 Da | 低通量高灵敏度方法(如X射线)用小分子端,高通量方法(如SPR)用大分子端 |
重原子数(HAC) | 9–16 | 最优命中率区间;<9可能结合模式过于多变 |
cLogP | 0.0–2.0 | 避免亲脂性驱动的非特异结合 |
氢键供体(HBD) | ≤3 | |
氢键受体(HBA) | ≤3 | |
可旋转键 | 0–3 | 减少熵罚,利于结合模式固定 |
手性中心数量 | 0–1(最多2) | 始终以外消旋体筛选;光学纯体留待优化阶段 |
水溶性 | ≥5 mM(5% DMSO) | 以实测为准,计算值不可靠 |
溶液稳定性 | >24小时 | 在筛选条件下实测 |
9.2 库容量与多样性
参数 | 推荐范围 | 注释 |
|---|---|---|
库大小 | 500–3000个片段 | X射线/NMR等低通量方法用500–1000;SPR/生化法可用至3000+ |
多样性指标 | 基于距离的多样性算法 | 注意:算法不应系统性偏向大片段;需确保小片段的代表性 |
每种骨架类型的变体数 | ≥3–5个 | 便于发现初步SAR,并识别结构崩溃点 |
9.3 分子识别与药效团覆盖
- • 片段须包含多样化的极性结合基团,能覆盖蛋白热点常见的氢键识别模式;
- • 每种结合药效团(如酰胺供体-受体对、碱性氮-芳香环组合等)应以多种不同骨架表达;
- • 图6所示的两种典型结合药效团(β-分泌酶的脒基类药效团、蛋白激酶A中的供体-受体对)可作为库设计的参考模板。
9.4 形状多样性
- • 同时包含**2D(平面)与3D(立体)**形状片段;
- • 推荐使用主成分惯量(PMI)图评估形状覆盖度;
- • 对于难靶标(PPI、变构位点),适当提高3D片段比例。
9.5 合成可行性
要求 | 标准 |
|---|---|
合成步数 | 从商业可购起始原料出发,≤4步 |
生长向量数 | 每个片段至少具备多个可扩展位置(三维方向) |
类似物可得性 | 优先选择商业上有已知类似物的骨架 |
特殊考量 | 针对筛选技术设计:如含¹⁹F原子辅助NMR检测,含溴原子辅助X射线晶体学相位求解 |
9.6 必须排除的化合物类型
类型 | 示例 | 排除理由 |
|---|---|---|
PAINS | 醌类、米迦勒受体、氧化还原循环化合物(如毒黄素) | 与蛋白质共价反应或干扰检测信号,在多种assay中均呈"活性" |
聚集体形成化合物 | 部分两亲性化合物 | 在高浓度筛选下形成胶束,产生假阳性 |
高活性官能团 | 醛、活泼酯、迈克尔受体、表氧化物等 | 非特异性共价修饰蛋白质 |
PrATs(泛活性2-氨基噻唑) | 2-氨基噻唑类 | 在广谱assay中频繁命中;注意:此类骨架并非绝对不可用,已有上市药物含此结构,需结合具体情况判断 |
十、FBDD的成功实践:典型案例回顾
10.1 已获批上市的FBDD药物
药物 | 靶标 | 适应症 | 关键FBDD机构 |
|---|---|---|---|
Vemurafenib | B-RAF V600E | 黑色素瘤 | Plexxikon/Roche |
Venetoclax | BCL-2 | 慢性淋巴细胞白血病 | AbbVie/Genentech(片段起点) |
截至2016年,已有30余个FBDD来源的化合物进入临床试验。
10.2 突破难靶标的代表性案例
BCL-xL/BCL-2(PPI靶标): AbbVie(原雅培)SAR by NMR技术的里程碑,从BCL-xL的片段苗头物出发,历经多轮优化,最终得到Navitoclax,进一步选择性优化为BCL-2选择性抑制剂Venetoclax(FDA 2016年批准)。
Ras("不可成药"靶标): Genentech与Vanderbilt大学分别通过NMR筛选独立发现了能结合Ras表面口袋、阻断SOS交换因子结合的片段,为K-Ras靶向药物的早期探索提供了起点。
cIAP1/XIAP(弱结合PPI): Astex在10 mM浓度下NMR筛选发现极弱苗头物,以结构指导化学优化,最终获得双靶标纳摩尔级活性抑制剂。
HCV NS3(变构机制): Astex通过片段筛选发现结合于蛋白酶-解旋酶结构域间界面的片段,揭示了全新变构调控机制,优化至低纳摩尔效力并具备细胞活性。
K-Ras G12C(共价结合): UCSF Wells课题组利用Tethering技术针对含活化半胱氨酸的K-Ras突变体,验证了共价片段策略在"不可成药"靶标上的可行性(为后续Sotorasib的发现奠定基础)。
十一、对学术界的特别建议
文章最后,作者们针对学术背景的研究者给出了几点特别提示:
- 1. 从小而精的库起步:500–1000个高质量片段,远胜于5000个质量参差不齐的片段。库的质量比规模更关键;
- 2. 充分利用生物物理技术:许多大学已有NMR、SPR和晶体学设施,不必等待资金建立完整筛选平台;
- 3. 优先建立正交验证体系:哪怕只有两种技术,也要保留一种用于交叉验证;
- 4. 不要低估库管理成本:DMSO溶液降解、供应商样品质量问题是实际项目中最常见的"隐藏地雷";
- 5. 合理使用商业库:表5列出了30余家供应商的库信息,但购买前必须评估溶解度数据、纯度确认方式,并结合目标靶标选择适当的子集;
- 6. 与工业界建立合作:FBDD的成功高度依赖化学、结构生物学和生物物理学的交叉协作,学术界应积极推动跨学科团队建设。
总结
本文的核心贡献在于:将片段库设计从经验性操作提升为有理论基础、有实验验证、有具体参数的系统性方法论。
无论是分子复杂度的概率框架、正交筛选技术的统计支撑、结合模式保守性的结构证据,还是片段库日常管理的操作细节,都体现了作者们深厚的工业经验和严谨的科学思维。
对于希望在学术环境中系统开展FBDD的研究者而言,这篇综述不仅是技术指南,更是一份来自一线实践者的诚意之作。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-12,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号