J. Am. Chem. Soc. | 模块化三维合成平台助力基于片段的药物发现

J. Am. Chem. Soc. | 模块化三维合成平台助力基于片段的药物发现

DrugIntel
发布于 2026-04-13 18:03:31
发布于 2026-04-13 18:03:31

文献来源:Gomez-Angel, A. R.; Klein, H. F.; Yao, S. Y. et al. J. Am. Chem. Soc.2025, 147, 29292–29303. DOI:10.1021/jacs.5c08786 通讯作者:Peter O'Brien,英国约克大学化学系 合作机构:AstraZeneca Hit Discovery, Discovery Sciences
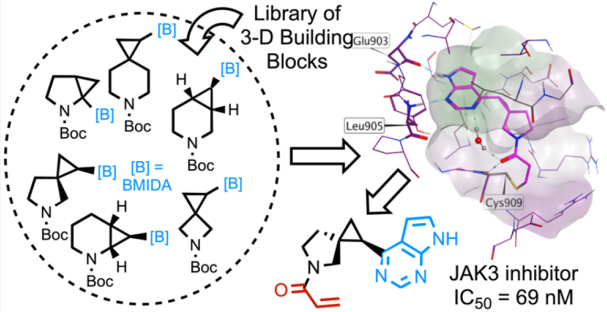
一、背景与问题:片段药物发现的"最后一公里"困境
1.1 FBDD 的崛起与成熟
基于片段的药物发现(Fragment-Based Drug Discovery, FBDD)自 20 世纪 90 年代兴起以来,已发展为制药工业中成熟且高效的先导发现策略。其基本逻辑在于:
- • 分子量小(MW 通常 < 300 Da),以极低浓度(通常 mM 级)对蛋白靶点进行弱结合筛查;
- • 化学空间覆盖高效:数千个片段即可有效覆盖相当于数百万化合物高通量筛选库所对应的化学空间;
- • 命中率高、起点质量好:片段命中物通常具备高配体效率(LE),便于后期优化。
迄今,FBDD 已推动 8 款药物上市,并产生超过 59 个临床候选分子,代表性药物包括 Venetoclax(BCL-2 抑制剂)、Erdafitinib(FGFR 抑制剂)等。
1.2 核心瓶颈:合成化学是片段生长的速率限制步骤
FBDD 流程可简化为三步:片段筛选 → 结构表征 → 片段生长/阐述(Fragment Elaboration)。前两步技术相对成熟,而第三步——将片段命中物"生长"为具有足够活性的先导化合物——长期是整个流程的速率决定步骤(rate-limiting step)。
这一瓶颈的具体表现包括:
问题类型 | 具体表现 |
|---|---|
合成可行性局限 | 片段生长往往依赖市售结构类似物,真正的向量扩展需从头开发新路线 |
工具箱同质化 | 药化工作者高度依赖 sp²–sp² C–C 偶联(如 Suzuki),导致分子趋于平面化 |
三维向量缺失 | 现有方法难以系统覆盖 sp³ 出射向量,三维化学空间探索受限 |
速度与项目节奏不匹配 | 新路线开发周期与药物发现项目推进速度不匹配 |
2016 年,Murray 与 Rees 在《Angew. Chem.》发出业界呼吁:
"需要在片段筛选前就建立好三维生长的方法学,从多个生长点/向量系统阐述片段。"
1.3 现有解决方案及其局限
此前已有若干应对策略:
- 1. 开发三维片段库(O'Brien 组等):需大量前期资源投入,通用性有限;
- 2. C–H 活化方法(Ley、Heightman 等):位点选择性与极性官能团耐受性仍是挑战;
- 3. Fragment Sociability概念(Murray/Rees 等):区分合成可及与不可及片段,但未提供系统解决方案。
二、核心策略:模块化三维合成平台的设计哲学
O'Brien 团队提出的解决方案独辟蹊径——不改变现有合成工具箱,而是设计一套全新的双功能三维构建块,使常规的药化反应自然产生三维结构多样性。
2.1 平台架构:两阶段闭环流程
阶段一:先导设计(Lead Design Phase)
片段命中物(X 射线确认结合模式)
↓
虚拟库枚举:片段 + 9 个三维构建块 + N-帽基组合
↓
RDKit 计算最低能量构象 → 出射向量分析
↓
计算对接(Docking)筛选候选先导分子阶段二:合成实施(Synthesis Phase)
溴代片段 + 三维构建块(BMIDA)
↓ Suzuki-Miyaura 偶联(已优化条件)
芳基化中间体(N-Boc 保护)
↓ HCl/二氧六环脱 Boc
游离胺
↓ 酰胺化 / 磺酰胺化 / 还原胺化 / Buchwald-Hartwig / SNAr
三维先导化合物关键优势:全部合成方法学在筛选前完成建立,命中物一旦确认即可快速按平台流程推进,无需临时开发新路线。
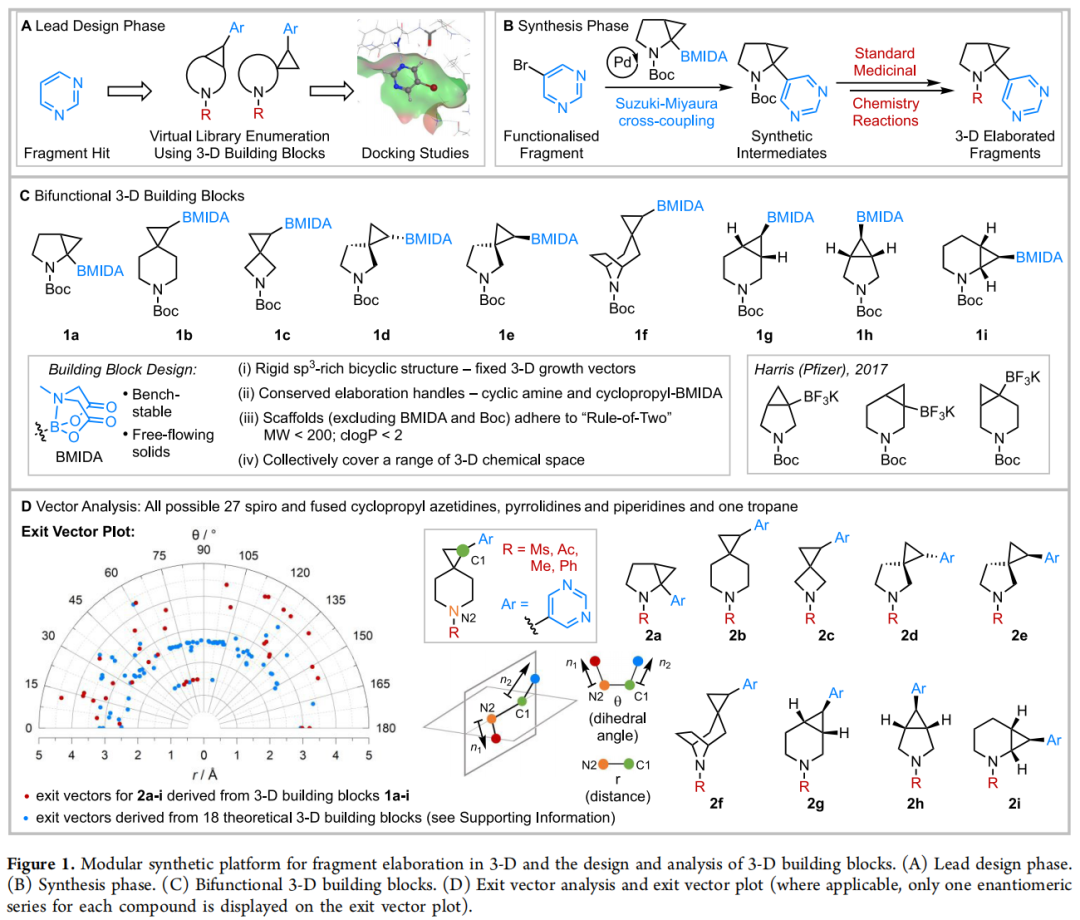
2.2 九个双功能三维构建块(1a–1i)的设计准则
每个构建块同时满足以下四项设计标准:
标准一:刚性 sp³ 富双环骨架含环丙烷
构建块采用稠合(fused)或螺环(spirocyclic)双环结构,核心均包含一个环丙烷单元。环丙烷在这里扮演关键角色:
- • 提供刚性三维构型,固定片段与帽基之间的空间向量关系;
- • 环丙烷在药物分子中广泛存在(已有充分的药代动力学数据积累);
- • sp³ 富骨架提升分子三维性(Fsp³),与更优的成药性相关。
标准二:两个正交保护的合成句柄
每个构建块均配备:
- • Boc 保护的环状胺(azetidine / pyrrolidine / piperidine 之一):可正交脱保护,接入 N-官能化帽基;
- • 环丙基 MIDA 硼酸酯(cyclopropyl BMIDA):用于 Suzuki-Miyaura 偶联连接片段。
MIDA 硼酸酯的战略价值:
- • 相比硼酸(boronic acid),BMIDA 空气稳定、可自由流动的结晶固体,易于纯化与储存;
- • Burke 课题组已证明 BMIDA 是理想的"枢纽"构建块(linchpin building block),可缓慢释放活性硼酸参与偶联;
- • 大量片段库以芳基/杂芳基溴化物形式存在(包括 FragLite 筛选集),与 BMIDA 的偶联有坚实文献基础。
标准三:符合 AstraZeneca "Rule-of-Two" 药用构建块规则
骨架部分(不含 BMIDA 和 Boc)须满足:
- • MW < 200 Da
- • clogP < 2
保证构建块本身的理化属性留有足够余量,与片段和帽基组合后仍处于先导化合物空间。
标准四:集体覆盖宽广的三维化学空间
通过 Grygorenko 出射向量图(Exit Vector Plot) 分析——以变化点之间的空间距离(r)和二面角(θ)为坐标——验证九个构建块共同覆盖宽广且分散的三维向量空间(r = 1.5–4.4 Å,θ 大范围分布)。
三、合成化学:三条路线实现克级制备
3.1 路线一:α-锂化/捕获策略(构建块 1a)
适用于稠合 2,3-吡咯烷-环丙烷 BMIDA(1a)的合成:
4-氯哌啶(3)
↓ s-BuLi / TMEDA, −78 °C → α-锂化
↓ 分子内环丙烷化 → azabicyclo[3.1.0]hexane 骨架
↓ 第二次 α-锂化 + (MeO)₃B → 硼酸(4)
↓ MIDA / HC(OEt)₃, DMSO, 100 °C, 48 h → 1a(43%,4.2 g/批)立体化学控制:将 MIDA 替换为 Burke 的手性等价物 BIDA(N-2-苄氧基环戊基亚氨基二乙酸),可将外消旋的 1a 分离为两个非对映异构体 1a'(26%) 和 1a''(20%),实现对映体纯构建块的制备,并通过转化为已知构型的偶联产物完成绝对构型归属。
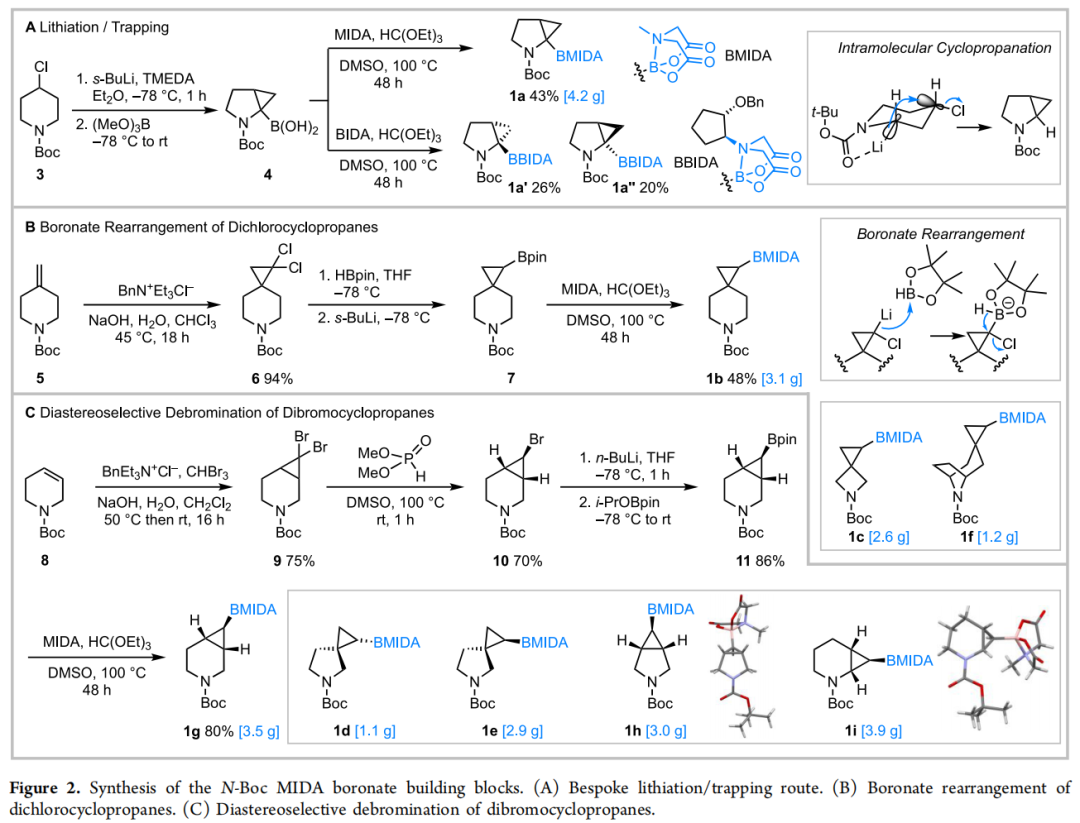
3.2 路线二:gem-二氯环丙烷硼酸酯重排策略(构建块 1b、1c、1f)
适用于螺环三维构建块的通用路线:
含外环烯烃的 N-Boc 环胺
↓ BnN⁺Et₃Cl⁻ / NaOH / CHCl₃, 相转移催化 → gem-二氯环丙烷(94%)
↓ s-BuLi / HBpin, −78 °C
(机制:Li-卤素交换 → HBpin 捕获 → 1,2-氢负离子迁移)
→ 环丙基频哪醇硼酸酯(Bpin)
↓ MIDA / HC(OEt)₃ → 构建块 1b(48%,3.1 g/批)亮点:热带骨架构建块 1f 经高度非对映选择性的二氯卡宾加成反应(二氯卡宾选择性从桥的对侧加成),以单一非对映异构体获得。
3.3 路线三:gem-二溴环丙烷非对映选择性单去溴策略(构建块 1d、1e、1g、1h、1i)
适用于稠合三维构建块的通用路线:
N-Boc 四氢吡啶(8)
↓ BnEt₃N⁺Cl⁻ / NaOH / CHBr₃, CH₂Cl₂–水 → gem-二溴环丙烷(9,75%)
↓ (MeO)₂P(O)H / KOtBu, DMSO, 100 °C
(Meijs-Doyle 单去溴,1985 年报道的欠开发反应)
→ exo-单溴环丙烷(10,70%)
(立体选择性源于:单溴环丙基碳负离子的质子化在位阻较小的 exo 面发生)
↓ n-BuLi / i-PrOBpin, −78 °C(立体保留的 Li-卤素交换)→ Bpin(11,86%)
↓ MIDA → 构建块 1g(80%,3.5 g/批)注意事项:对于螺环吡咯烷系列(1d vs. 1e),由于环丙烷两侧位阻相近,单去溴反应缺乏非对映选择性(55:45),需通过分离非对映体分别获得 1d 和 1e,构型通过中间体 X 射线晶体学归属。
所有 9 个构建块现已商业化供应,便于领域内快速推广。
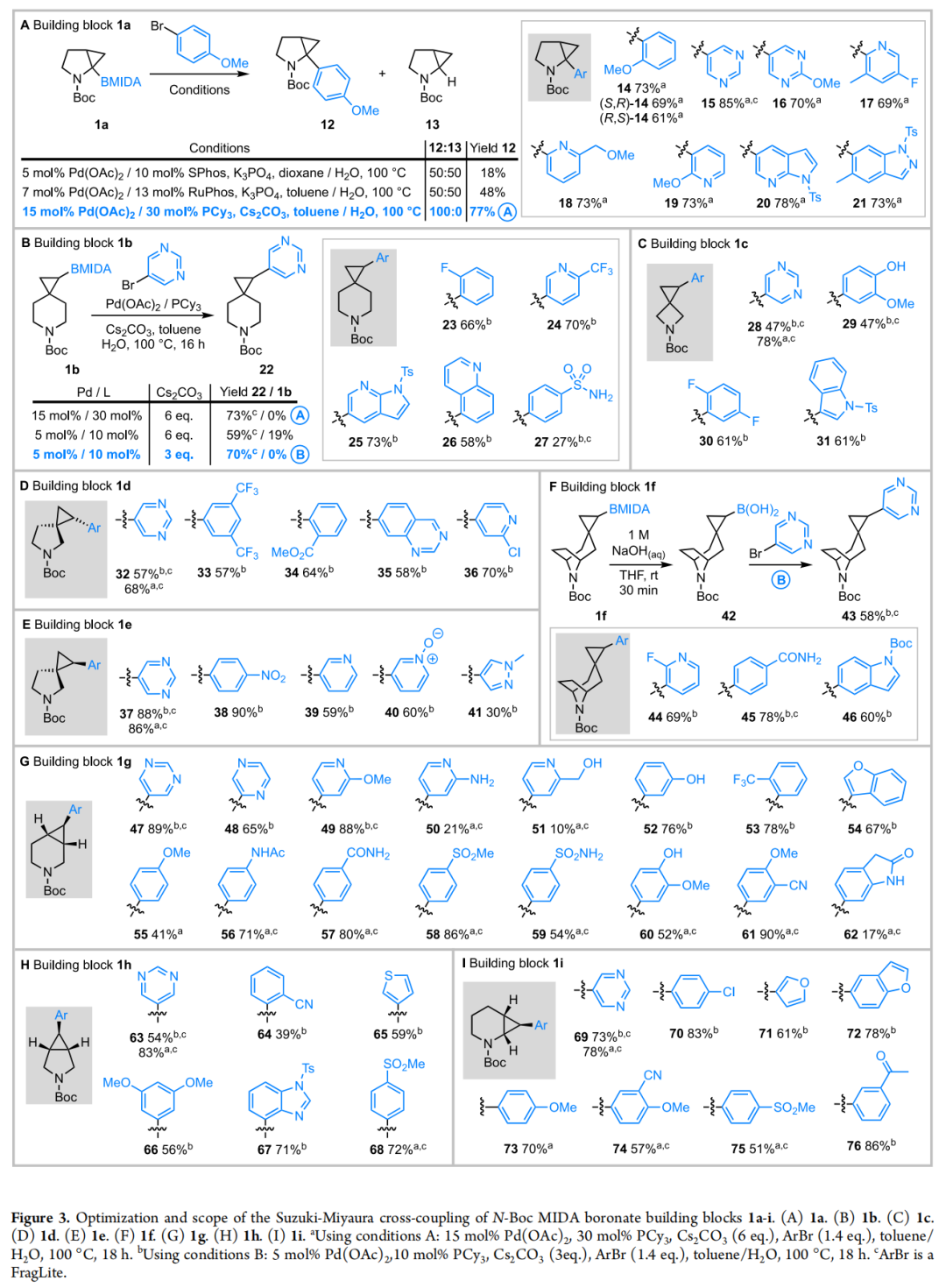
四、方法学验证:65 例 Suzuki-Miyaura 偶联的系统研究
4.1 条件优化:攻克环丙基 BMIDA 偶联的难题
环丙基 BMIDA 与普通芳基 MIDA 硼酸酯相比,偶联更具挑战性——在原位水解产生环丙基硼酸后,后者更容易发生原去硼化(protodeboronation),生成无取代的环丙烷副产物(13)。
系统条件筛选结果:
条件 | Pd 源/配体 | 碱/溶剂 | 12:13 比例 | 12 产率 |
|---|---|---|---|---|
Burke 条件 | Pd(OAc)₂/SPhos(5/10 mol%) | K₃PO₄, 二氧六环/水 | 50:50 | 18% |
RuPhos 条件 | Pd(OAc)₂/RuPhos(7/13 mol%) | K₂CO₃, 甲苯/水 | 50:50 | 48% |
条件 A(优化) | Pd(OAc)₂/PCy₃(15/30 mol%) | Cs₂CO₃(6 eq.),甲苯/水 | >99:1 | 77% |
条件 B(常规) | Pd(OAc)₂/PCy₃(5/10 mol%) | Cs₂CO₃(3 eq.),甲苯/水 | ~95:5 | 59–73% |
条件 A 适用于难偶联底物(如 1a);条件 B 适用于多数构建块(大幅降低钯用量,更具实用性)。
特殊情况——热带构建块 1f:由于 1f 在甲苯/水体系中溶解性差,先用 NaOH(aq) 原位水解 MIDA 硼酸酯生成假定的硼酸(42),再进行偶联,以 58–78% 收率获得目标产物(44–46)。
4.2 底物适用范围
63 例偶联涵盖 46 种不同芳基/杂芳基溴化物:
- • 电子效应:吸电子(硝基、三氟甲基、磺酰甲基等)与富电子(甲氧基、氨基等)芳基溴均兼容;
- • 杂环多样性:嘧啶、吡啶、氮杂吲哚、吲唑、喹啉、喹唑啉、吡唑、苯并呋喃、噻吩、苯并咪唑、呋喃等;
- • 产率范围:10–90%,多数 ≥ 60%;
- • FragLite 兼容:11 种不同的 FragLite 分子(高极性、富挑战性的芳基溴/碘化物筛选集)均成功偶联;
- • 立体化学:X 射线晶体学证实偶联过程保持构型。
五、先导化合物合成验证:32 例三维先导化合物
5.1 N-官能化多样化
脱除 Boc 后,以常规药化工具箱完成多样化:
反应类型 | 代表产物 | 功能意义 |
|---|---|---|
磺酰胺化 | 77, 86–94 | 提供氢键供/受体,系列用于三维出射向量 X 射线分析 |
酰胺化(酰氯/T3P) | 79, 81, 83 | 丙烯酰胺(79)作为共价弹头 |
还原胺化 | 80 | N-烷基化,引入 sp³ 多样性 |
Buchwald-Hartwig | 82 | N-芳基化,拓展化学空间 |
SNAr | 85 | 直接 N-芳基化,无需金属催化 |
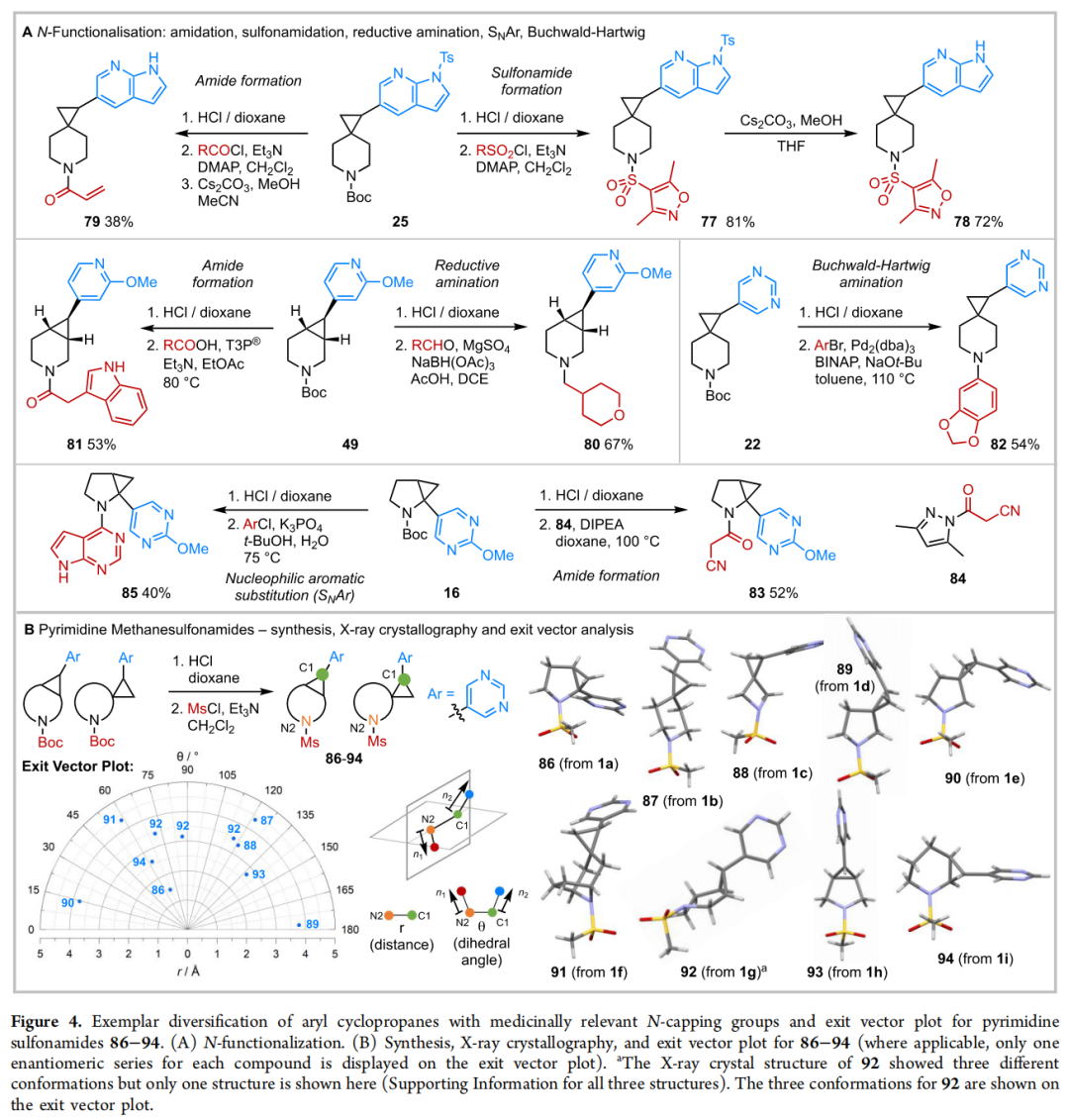
5.2 三维出射向量的实验验证
以 5-溴嘧啶为共同片段,将 9 个构建块(1a–1i)分别偶联并转化为相应的甲磺酰胺衍生物(86–94),进行 X 射线单晶衍射分析。
- • 所有 9 个化合物均为结晶固体,成功解析晶体结构;
- • 出射向量图(r vs θ)证实 86–94 覆盖宽广且彼此区分的三维化学空间;
- • 化合物 92(源于 1g)在不对称单元中发现三种构象,均呈现于向量图中。
5.3 理化及 DMPK 性质分析
理化性质(32 个先导化合物):
指标 | 均值 | Churcher 先导化合物空间标准 |
|---|---|---|
clogP | 0.46 | −1 < clogP < 3 |
MW(Da) | 266 | 200–350 |
Fsp³ | 0.56 | 越高越优 |
达标率 | 29/32 | — |
DMPK Wave1 分析(12 个化合物,AstraZeneca):
指标 | 结果 |
|---|---|
测定 logD | −0.4 至 3.6 |
水溶性 | 49.0 至 >981 μM(全部合格) |
人肝微粒体(HLM)稳定性 | 10/12 化合物良好 |
大鼠肝细胞(RH)稳定性 | 10/12 化合物良好 |
代谢不稳定的 2 个化合物(78、81)均含有苄基位或富电子吲哚环,属于分子整体性质问题,与环丙烷构建块骨架本身无关——构建块无固有代谢缺陷。
六、概念验证:从假想二维片段到 69 nM 选择性 JAK3 抑制剂
6.1 靶点背景:JAK3 与 Ritlecitinib
Janus 激酶(JAK)家族包含 JAK1、JAK2、JAK3 和 TYK2,是细胞因子信号传导的核心调控者,也是自身免疫、自体炎症和过敏性疾病的重要治疗靶点。
Ritlecitinib(PF-06651600):
- • 2023 年 FDA 批准,用于治疗斑秃;
- • JAK3 选择性共价抑制剂(通过丙烯酰胺与 JAK3 特异性 Cys909 形成共价键);
- • 吡咯并嘧啶核心通过氢键与铰链区 Glu903 和 Leu905 结合;
- • 已有高分辨率 JAK3 共晶结构(PDB 可获)。
6.2 先导设计流程
- 1. 假设:将吡咯并嘧啶视为初始二维片段命中物,在 JAK3 铰链区以与 Ritlecitinib 相同的模式结合;
- 2. 虚拟库枚举:从吡咯并嘧啶的 4 位出发,与 9 个构建块及丙烯酰胺帽基组合,生成 9 个虚拟先导化合物;
- 3. 计算对接:以含/不含 Cys909 共价键两种方式对接,筛选评分最优且与 Ritlecitinib 构象相近的候选物;
- 4. 优选:化合物 95(源于 1b)和 96(源于 1e)进入合成验证阶段。
96 的对接模型(R,R 构型)显示其丙烯酰胺羰基能形成 Ritlecitinib 中被识别的水介导氢键(嘧啶 → 羰基),这一构象特征被认为是更优抑制活性的结构基础。
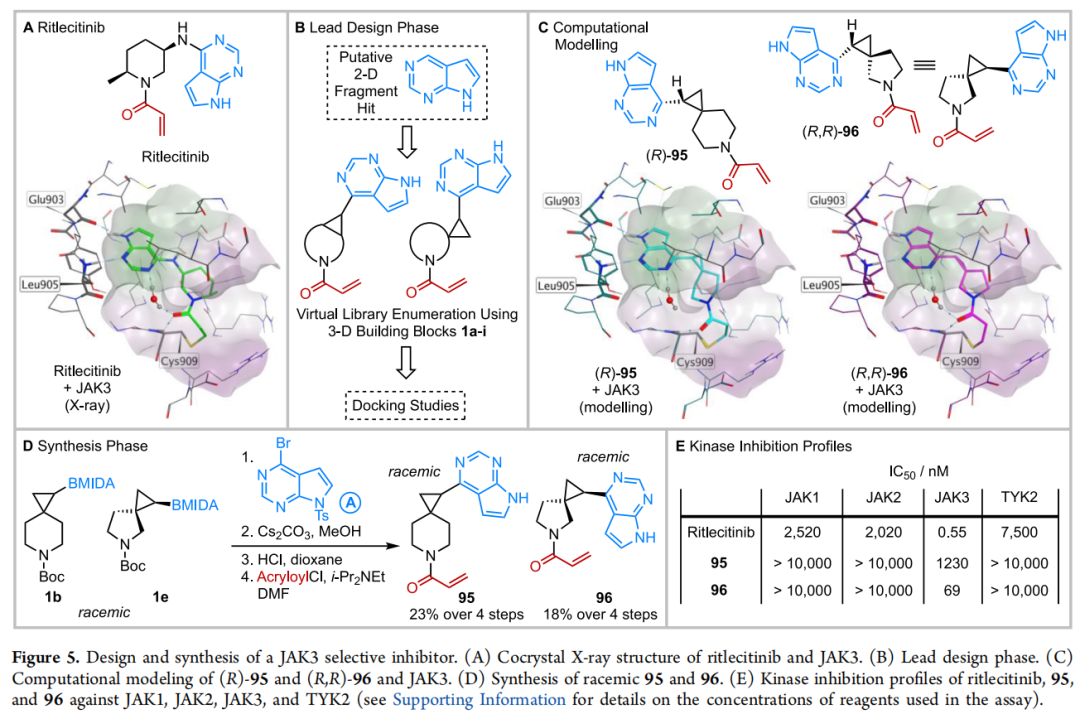
6.3 合成路线(4 步,已有先例)
化合物 95(源于 1b):
1b + 溴代吡咯并嘧啶 → Suzuki 偶联(条件 A)
→ Cs₂CO₃/MeOH 脱 N-Tos
→ HCl/二氧六环脱 Boc
→ 丙烯酰氯 / i-Pr₂NEt → 95(23%,4 步)化合物 96(源于 1e):
同上路线,使用 1e → 96(18%,4 步)6.4 生物活性评估
激酶抑制 IC₅₀(nM),1 h 固定时间点法:
化合物 | JAK1 | JAK2 | JAK3 | TYK2 |
|---|---|---|---|---|
Ritlecitinib | 2,520 | 2,020 | 0.55 | 7,500 |
95 | >10,000 | >10,000 | 1,230 | >10,000 |
96 | >10,000 | >10,000 | 69 | >10,000 |
关键结论:
- • 96 对 JAK3 的 IC₅₀ = 69 nM,对 JAK1、JAK2、TYK2 均无明显抑制(>10,000 nM);
- • 96 的 JAK3 选择性优于 Ritlecitinib(后者对 JAK1/JAK2 仍有 μM 级活性);
- • 96 为外消旋体,Ritlecitinib 为单一对映体,单一活性对映体的活性有望进一步提升;
- • 谷胱甘肽(GSH)反应性:96 的 t₁/₂ = 1254 min,Ritlecitinib 为 2020 min(相近量级),96 的共价弹头活性适中,符合选择性共价抑制剂设计原则。
七、方法论意义与学术价值
7.1 对 FBDD 领域的贡献
维度 | 贡献 |
|---|---|
概念层面 | 首次提出"预建立方法学 + 模块化构建块"的闭环平台,系统性回应工业界的 FBDD 三维生长挑战 |
合成层面 | 开发三条通用合成路线,覆盖稠合/螺环/热带骨架的 BMIDA 构建块克级制备 |
信息层面 | 系统性出射向量分析(27 种虚拟构建块,112 个虚拟先导化合物),提供三维化学空间地图 |
应用层面 | JAK3 选择性抑制剂(IC₅₀ = 69 nM)的快速发现,直观展示平台加速能力 |
推广层面 | 9 个构建块商业化,合成条件全部使用常规药化反应,大幅降低应用门槛 |
7.2 与相关工作的对比定位
- • 相比 Harris et al.(Pfizer,2017):后者开发了类似的稠合环丙基吡咯烷/哌啶 BF₃K 盐,但并非针对片段阐述设计,缺乏系统性的三维向量分析和 FBDD 应用框架;
- • 相比 Grygorenko 等:后者提供了 gem-二氟环丙烷类似物,但缺乏 BMIDA 双功能设计;
- • 相比 Gutiérrez-Bonet/Merck(2022):后者实现了环丙基硼酸酯的不对称合成,与本工作互补(本工作先完成外消旋路线),为未来对映体纯版本提供参考。
7.3 局限性与未来方向
- • 当前覆盖范围:9 个构建块(出射向量图中的红色点)仅覆盖 27 个理论构建块的一部分,仍有蓝色区域(18 个理论构建块)未被实验验证;
- • 对映体纯化:主系列构建块为外消旋体,BIDA 路线实现了对映体纯,但需进一步推广至其他骨架;
- • 生长方向限制:平台以溴代片段为起点,不含溴取代位点的片段命中物需要额外策略(如 C–H 活化);
- • 未来方向:团队明确表示将扩展构建块集合,填补三维向量/化学空间的空白。
八、总结
O'Brien 课题组与 AstraZeneca 的合作成果代表了片段药物发现合成方法学领域的一次系统性突破:通过预建立合成方法学 + 模块化双功能三维构建块的组合,将片段生长的速率限制步骤从"合成路线开发"转变为"按图索骥的执行"。
平台的核心优势在于其可编程性——虚拟库枚举、计算对接与已验证的合成路线三者构成一个可重复执行的闭环,使得从二维片段命中物到三维先导化合物的转化可以像"流水线"一样运作。JAK3 抑制剂(IC₅₀ = 69 nM,JAK3 选择性)的发现,不仅是化学方法学的概念验证,也展示了其直接转化为药物研究价值的潜力。
随着构建块的商业化和平台的进一步扩展,这一工具集有望成为 FBDD 领域的基础设施之一。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-13,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号