虚拟筛选之化合物批量处理
原创
虚拟筛选之化合物批量处理
原创
追风少年i
发布于 2026-04-15 10:23:03
发布于 2026-04-15 10:23:03
作者,Evil Genius
大家想做科研么?如果致力于科研,努力读博有个编制吧,反正以我的经验而言,有个保底才行。
我们无法同时拥有青春和成熟的心态,无论什么情况下,大家都需要一张底牌来面对以后的未知,而编制,就是大家最大的底牌,哪怕这张底牌,每个月只能带来3000的收入。
像我一样没有底牌的人,抗风险能力真的有点低。
今天我们来完成虚拟筛选的化合物(配体分子)准备。
目前化合物分子存储的主要格式
类别 | 格式名称 (扩展名) | 核心特点与存储内容 | 主要应用场景 | 优点 | 缺点 |
|---|---|---|---|---|---|
文本序列格式 | SMILES (.smi 或 .txt) | 用简短的ASCII字符串表示分子的二维连接信息(原子、键、分支、环)。示例:CC(=O)Oc1ccccc1C(=O)O (阿司匹林) | 数据库存储、文档记录、快速检索、机器学习模型输入。 | 极其简洁,人类可读,存储空间小。 | 不包含三维坐标,不同写法可能对应同一分子(非绝对唯一)。 |
InChI/InChIKey (.txt) | IUPAC制定的国际标准,分层编码分子式、连接、立体化学、电荷等所有信息。InChIKey是其固定长度的哈希版本。 | 数据库索引、文献检索、唯一性标识。 | 绝对唯一,标准开放,能完整描述分子结构。 | 字符串很长,不包含三维坐标。 | |
SMARTS (.smar) | SMILES的扩展,用于定义分子子结构模式。 | 子结构搜索(如查找所有芳香环)、药物设计中的骨架筛选。 | 强大的模式匹配能力,是化学信息学搜索的核心工具之一。 | 语法比SMILES更复杂,专用于查询。 | |
三维坐标文件格式 | MOL/SDF (.mol, .sdf) | MDL/Biovia开发的行业标准。.mol 存储单个分子;.sdf 可在一个文件里存储多个分子及各自属性数据(如logP、活性值)。 | 分子结构可视化、分子对接、虚拟筛选、化学信息学软件通用交换格式。 | 同时存储原子坐标和连接表,信息完整;.sdf 便于管理化合物库。 | 有V2000(限制999原子)和V3000(支持大数据)版本差异,存在兼容性问题。 |
MOL2 (.mol2) | Tripos公司开发,除坐标和连接外,明确包含原子类型(如O.3, C.ar)和部分原子电荷。 | 分子力学计算、分子对接(如AutoDock)、力场参数化。 | 信息丰富,明确指定了杂化状态和电荷,避免软件误判,可靠性高。 | 文件体积较大。 | |
PDB (.pdb) | 蛋白质数据库的原始格式,用于存储生物大分子(蛋白、核酸)和小分子的三维结构。 | 结构生物学、蛋白质-配体复合物分析、分子对接。 | 生物大分子研究的基石,全球通用。 | 格式规范严格(列固定),易出错,不推荐用于存储小分子。 | |
XYZ (.xyz) | 最简单的格式:第一行是原子数,第二行是注释,后面每行是元素符号和X、Y、Z坐标。 | 量子化学计算(Gaussian, ORCA)、分子动力学轨迹、分子可视化入门。 | 极度简单,任何文本编辑器可写,易解析。 | 不含连接信息,软件需自行猜测成键,可能导致错误;信息量最少。 | |
其他专门格式 | CIF (.cif) | 晶体学信息文件,精确存储晶体结构、空间群、晶胞参数、原子位置及占有率。 | X射线晶体学、材料科学、固体化学。 | 描述晶体结构的国际标准,信息完整且高度精确。 | 不适用于溶液中的单个小分子模型。 |
FASTA (.fasta 或 .fa) | 生物信息学标准,用单字母代码表示氨基酸(蛋白质)或碱基(DNA/RNA)序列。 | 生物序列比对(BLAST)、数据库搜索、进化分析。 | 序列存储的绝对标准,格式简单,支持模糊碱基。 | 只含一维序列信息,不含三维结构。 |
格式的选择也很简单
📊 只需要记录分子结构、建数据库或写论文:首选 SMILES 或 InChI。
👀 需要看分子的3D形状、做分子对接或虚拟筛选:首选 SDF (.sdf) 或 MOL2 (.mol2)。
🧬 研究对象是蛋白质、DNA,或做分子对接需要蛋白结构:必须使用 PDB (.pdb)。
⚛️ 自己做量子化学计算,只关心原子坐标:XYZ (.xyz) 格式最方便。
💎 分析的是晶体(如MOF、沸石):必须使用 CIF (.cif)。
我们目前做虚拟对接,需要配体分子的三维结构,所以一般就是sdf或者mol2格式。
小分子结构(数据库):采用moI2和mol(sdf)的存储格式 生物大分子(蛋白质、核酸)结构:采用moI2和pdb的存储格式。这些结构文件格式可以被大多数分子模拟软件所识别。
mol2格式文献
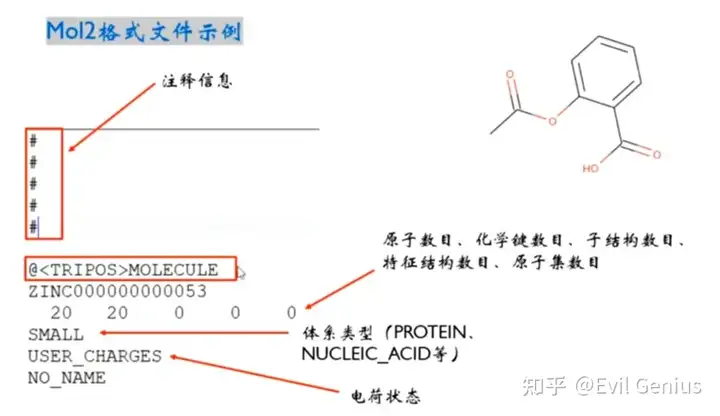
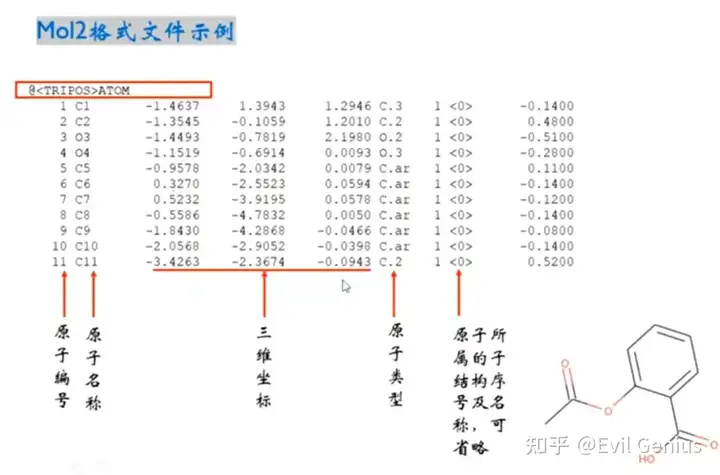
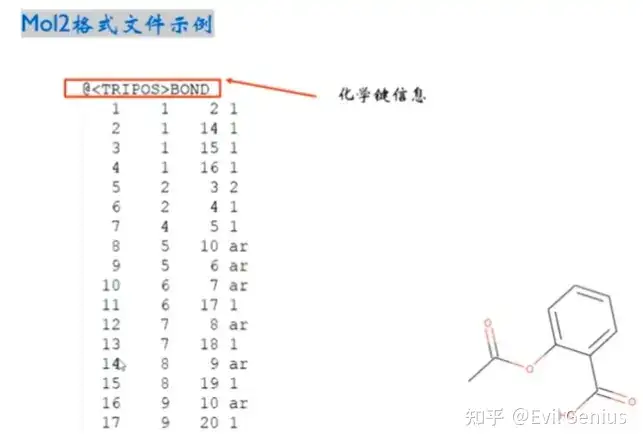
其中化学键的类型与数字标记
数字标记 | 字符串标记 | 化学键类型 | 说明 |
|---|---|---|---|
1 | single | 单键 | 最常见的键类型,表示一对共用电子。 |
2 | double | 双键 | 包含两对共用电子。 |
3 | triple | 三键 | 包含三对共用电子。 |
4 | ar 或 aromatic | 芳香键 | 用于苯环等芳香体系,键级在1和2之间,具有离域特性。 |
5 | am 或 amide | 酰胺键 | 特指蛋白质或多肽中连接氨基酸的酰胺/肽键(-CO-NH-)。 |
6 | du | 虚键 | 用于表示分子间的相互作用(如氢键)或暂定的连接关系。 |
7 | un | 未知键 | 当无法确定或识别化学键的具体类型时使用。 |
8 | nc | 不相连 | 明确标记两个原子之间没有化学键,主要用于避免自动补键算法产生错误连接。 |
- | quadruple | 四键 | 非常罕见的键型,主要存在于某些过渡金属二聚体(如Re-Re键)中 |
我们通常在数据库下载的是sdf的2D坐标,对接的时候需要转化成3D坐标。
接下来就是转化的方法,Open Bable。
Open Babel是一款开源自由软件,使用Open Babel可以将一种化学结构类型的文件格式转换成另一种文件格式,非常方便的进行各种类型的化学结构文件进行相互转换。
虚拟筛选离不开分子对接,分子对接离不开分子文件,分子文件的处理离不开openbabel。
这个软件开源,不需要收取任何费用,支持win以及linux。
支持SMILES,PDB,MOL2,SDF等等110种化学分子格式文件(pdbqt不建议使用此软件转化),其中绝大部分都是可以相互(批量)转换的,例如|维的SMILES转化为2维或者3维的MOL2等等,加全氢原子,或者极性氢原子,分子能量最小化处理。同时可以计算分子指纹,比较分子相似度。
我们来看一下使用方法
转化格式:
obabel -imol2 ligand.mol2 -opdb -O ligand.pdb-imol2:输入结构文件的格式
-opdb:输出结构文件的格式
-O:输出文件的文件名(选择性输入),obabel会自动识别文件扩展名。
--gen3d:生成3D构像一般用于smile等只有2D结构的文件,转换为3D带有空间结构的构象。
-m:一般用于被转换的文件中有多个分子。转换后文件将这些分子单独保存(虚拟筛选经常使用)。
加氢
-h:表示加全氢,不考虑质子化状态
-p<pH>:表示加指定H的氢(-h,-p参数只能用一个,参数相互冲突)
我们来看看所有参数
obabel -H
Usage:
obabel[-i<input-type>] <infilename> [-o<output-type>] -O<outfilename> [Options]
The extension of a file decides the format, unless it is overridden
by -i or -o options, e.g. -icml, or -o smi
See below for available format-types, which are the same as the
file extensions and are case independent.
If no input or output file is given stdin or stdout are used instead.
More than one input file can be specified and their names can contain
wildcard chars (* and ?). The format of each file can be different unless
the -i option has been used, when they are all the same.
By default, the molecules are aggregated in the output file,
but see -m option, Splitting, below.
Options, other than -i -o -O -m, must come after the input files.
Conversion options
-f <#> Start import at molecule # specified
-l <#> End import at molecule # specified
-e Continue with next object after error, if possible
-z Compress the output with gzip
-zin Decompress the input with gzip
-k Attempt to translate keywords
-H Outputs this help text
-Hxxx (xxx is file format ID e.g. -Hcml) gives format info
-Hall Outputs details of all formats
-V Outputs version number
-L <category> Lists plugin classes of this category, e.g. <formats>
Use just -L for a list of plugin categories.
Use -L <ID> e.g. -L sdf for details of a format or other plugin.
-m Produces multiple output files, to allow:
Splitting: e.g. obabel infile.mol -O new.smi -m
puts each molecule into new1.smi new2.smi etc
Batch conversion: e.g. obabel *.mol -osmi -m
converts each input file to a .smi file
Interface to OBAPI internals
API options, e.g. ---errorlevel 2
errorlevel # min warning level displayed
To see a list of recognized file formats use
babel -L formats [read] [write]
To see details and specific options for a particular format, e.g CML, use
babel -L cml我们先来转化一个分子
2D分子文件
2244
-OEChem-04132606022D
21 21 0 0 0 0 0 0 0999 V2000
3.7321 -0.0600 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
6.3301 1.4400 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
4.5981 1.4400 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
2.8660 -1.5600 0.0000 O 0 0 0 0 0 0 0 0 0 0 0 0
4.5981 -0.5600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
5.4641 -0.0600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
4.5981 -1.5600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
6.3301 -0.5600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
5.4641 -2.0600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
6.3301 -1.5600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
5.4641 0.9400 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.8660 -0.5600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
2.0000 -0.0600 0.0000 C 0 0 0 0 0 0 0 0 0 0 0 0
4.0611 -1.8700 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
6.8671 -0.2500 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
5.4641 -2.6800 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
6.8671 -1.8700 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
2.3100 0.4769 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.4631 0.2500 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
1.6900 -0.5969 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0
6.3301 2.0600 0.0000 H 0 0 0 0 0 0 0 0 0 0 0 0格式转化
obabel -isdf test.sdf -opdb -O test.pdb --gen3d -p 7.4看一下输出的文件
COMPND 2244
AUTHOR GENERATED BY OPEN BABEL 3.1.0
HETATM 1 O UNL 1 1.108 -0.092 -0.004 1.00 0.00 O
HETATM 2 O UNL 1 1.737 0.314 2.611 1.00 0.00 O1-
HETATM 3 O UNL 1 2.218 -1.702 3.508 1.00 0.00 O
HETATM 4 O UNL 1 1.043 -0.824 -2.233 1.00 0.00 O
HETATM 5 C UNL 1 0.284 -1.114 0.527 1.00 0.00 C
HETATM 6 C UNL 1 0.538 -1.578 1.829 1.00 0.00 C
HETATM 7 C UNL 1 -0.805 -1.619 -0.190 1.00 0.00 C
HETATM 8 C UNL 1 -0.251 -2.609 2.363 1.00 0.00 C
HETATM 9 C UNL 1 -1.596 -2.629 0.360 1.00 0.00 C
HETATM 10 C UNL 1 -1.314 -3.134 1.627 1.00 0.00 C
HETATM 11 C UNL 1 1.600 -1.026 2.707 1.00 0.00 C
HETATM 12 C UNL 1 1.419 -0.043 -1.370 1.00 0.00 C
HETATM 13 C UNL 1 2.317 1.129 -1.636 1.00 0.00 C
HETATM 14 H UNL 1 -1.024 -1.244 -1.126 1.00 0.00 H
HETATM 15 H UNL 1 -0.043 -2.980 3.303 1.00 0.00 H
HETATM 16 H UNL 1 -2.394 -3.004 -0.176 1.00 0.00 H
HETATM 17 H UNL 1 -1.889 -3.895 2.020 1.00 0.00 H
HETATM 18 H UNL 1 2.498 1.654 -0.722 1.00 0.00 H
HETATM 19 H UNL 1 1.848 1.786 -2.338 1.00 0.00 H
HETATM 20 H UNL 1 3.246 0.782 -2.038 1.00 0.00 H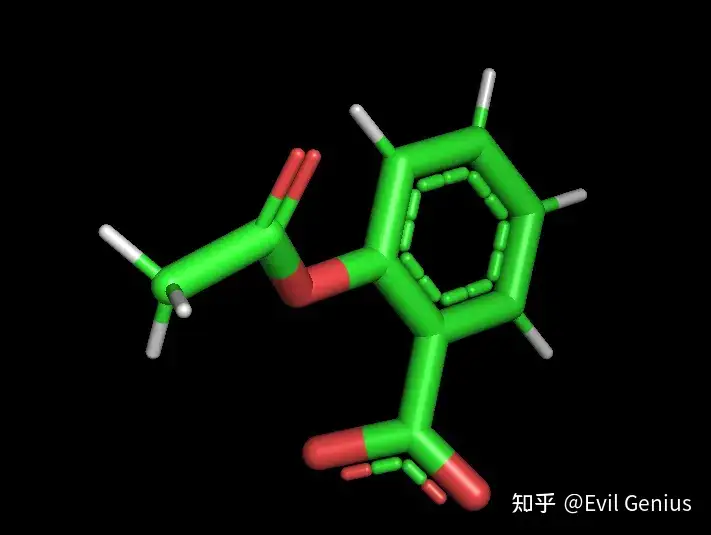
接下来批量处理sdf文件,拆分成每个分子。
obabel -isdf PubChem_search_records.sdf -opdb -O ./.pdb -m
我们随便看一个
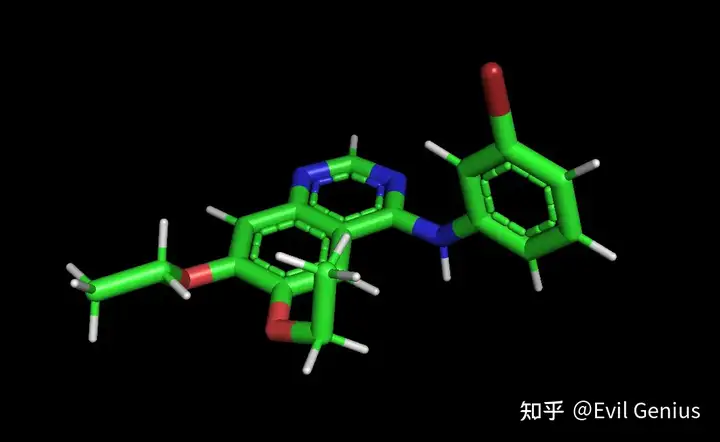
这样化合物数据库就准备好了,虚拟筛选即可
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号