文献分享(Stereo-seq)--一种保守的再上皮化程序是胰腺导管腺癌恶性转化的基础
原创
文献分享(Stereo-seq)--一种保守的再上皮化程序是胰腺导管腺癌恶性转化的基础
原创
追风少年i
发布于 2026-05-06 10:16:34
发布于 2026-05-06 10:16:34
作者,Evil Genius
假期结束,牛马上线。
无论什么时候,希望大家都保持乐观的心态,30岁以后确实和20多岁很不一样,有了家庭孩子,事业各方面都容易受挫,磕磕绊绊;生活压力也大了很多,回想工作以后,最快乐的时候就是刚参加工作的时候,住的360块一个月的房子,工资4000多,只要养活自己就行。
现在就是好好做一做,坚持一天是一天,趁着自己还有学习的劲头,往自己身上加一些技能,至于有没有用,就让命运来决定吧。
今天我们分享华大空转(Stereo-seq)的文献,通讯作者是外国人,单位是清华大学。

大家还记得之前给大家说过每个组学对基因的分类是有区别的么?大家还知道基因组是怎么划分基因的么?还有转录组的划分,希望大家心里有数,多组学的联合分析绝对是趋势。
知识积累
胰腺上皮内瘤变(PanIN)在健康个体中普遍存在,但其可进展为浸润性胰腺导管腺癌(PDAC),是PDAC最主要的癌前病变。
PDAC的恶性转化核心在于:PanIN细胞获得穿透BM、脱离导管、浸润实质组织的能力。
分子机制与动物模型
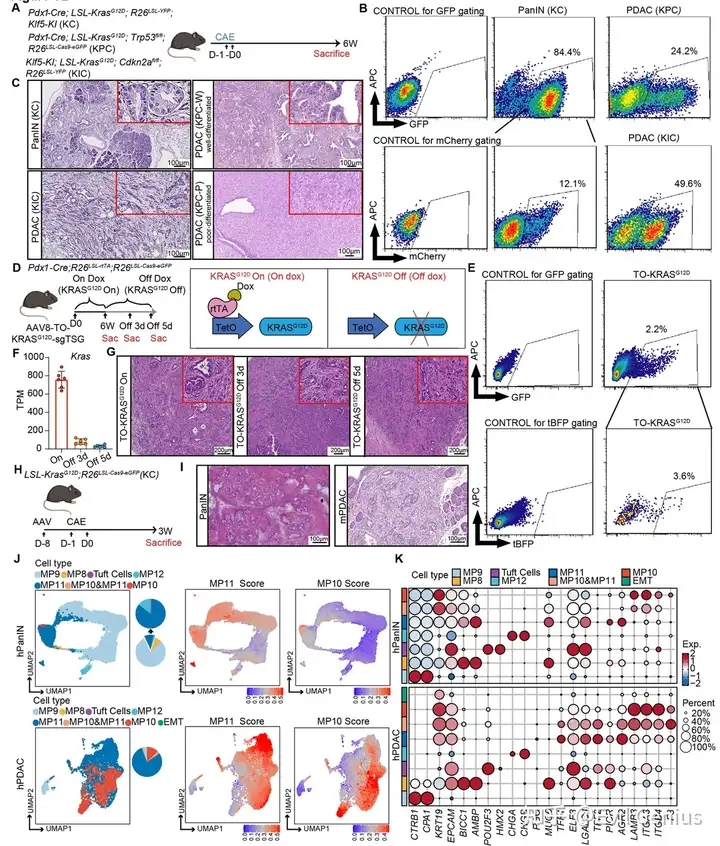
小鼠模型显示:Kras突变 + 炎症 共同诱导PanIN形成。
Kras突变“锁定”一种肿瘤样程序,来源于短暂存在的化生性祖细胞。
PanIN→PDAC进展需要肿瘤抑制基因(TSG) 的额外缺失(如Cdkn2a、Trp53)。
PDAC发生伴随显著的转录可塑性,但具体驱动恶性程序的基因尚不完全清楚。
恶性侵袭的不同模式
上皮-间充质转化(EMT):
抑制E-钙黏蛋白,失去极性,单个间充质样细胞播散。
在小鼠PDAC模型中存在,但在人PDAC中仅约16%的晚期肿瘤出现,并非必需机制。
集体侵袭:
由整合素粘附复合物(IAC) 驱动,包括局灶粘附(FA)和侵袭小体。
半桥粒(HD)与伤口愈合程序的意外联系
1型半桥粒(HD) 通常局限于皮肤和角膜,功能是将复层上皮附着于BM。
在皮肤伤口边缘的角质形成细胞中,HD组分显著上调,并参与再上皮化过程中的细胞迁移。
伤口诱导的角质形成细胞程序包含大量与上皮迁移相关的粘附、骨架、蛋白水解、代谢基因。
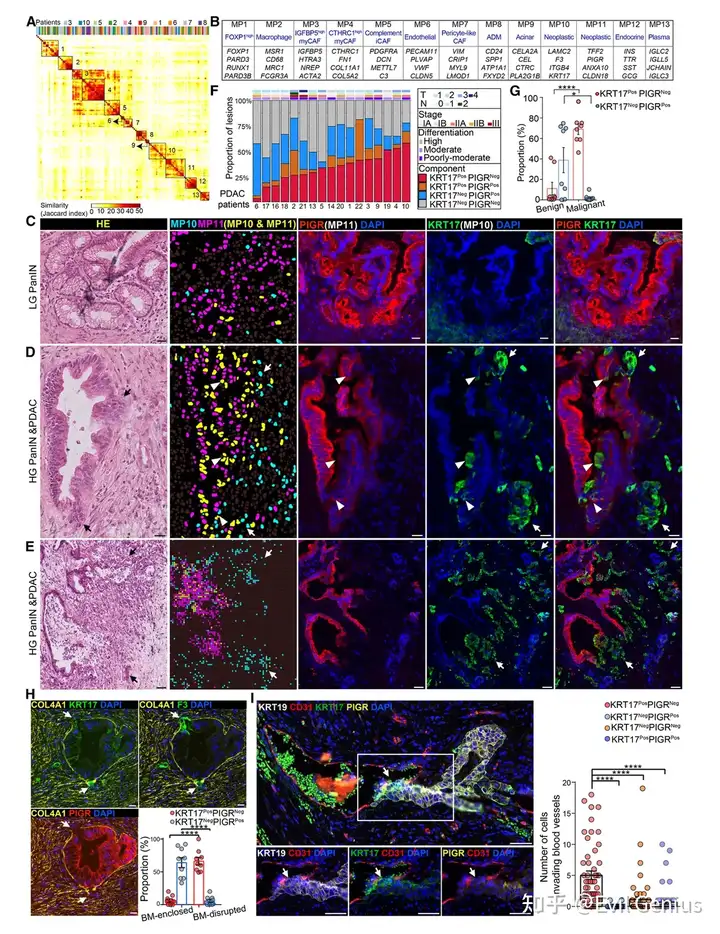
结果1、与人类胰腺导管癌恶性肿瘤相关的转录程序
研究目标与方法
目标:识别与PDAC恶性程度相关的基因表达程序及其空间分布。
样本:10例未经治疗的、手术切除的原发性PDAC。
技术:Stereo-seq空间转录组学 + 非负矩阵分解(NMF)+ 稳健排序聚合(RRA)。
元程序(MP)的鉴定与分类
共鉴定出13个元程序(MP),其中大多数对应非肿瘤细胞(如内分泌细胞、腺泡细胞、成纤维细胞、巨噬细胞等)。
仅MP10和MP11与肿瘤细胞相关:
MP11 → 对应经典型PDAC
MP10 → 对应基底样型PDAC(预后更差)
MP10高表达在所有PDAC队列中均强烈预示不良预后。
MP10与MP11在PanIN→PDCA进展中的动态变化
病变阶段 | MP表达模式 | 形态学特征 |
|---|---|---|
低级别PanIN(LG-PanIN) | 仅表达MP11 | 良性腺体形态 |
高级别PanIN(HG-PanIN) | MP11 + MP10 共表达(同一细胞) | 恶性克隆沿导管定植 |
浸润性癌细胞 | 仅表达MP10(MP11关闭) | 核异型性、腺体结构破坏 |
关键发现:MP10与恶性侵袭行为直接相关
基底膜突破:发生微浸润的细胞几乎总是从MP11转换为MP10。
血管浸润:参与血管浸润的癌细胞几乎全部只表达MP10,不表达MP11。
良性 vs 恶性:
MP11⁺only → 良性、结构规整的上皮
MP10⁺only → 恶性、浸润性癌细胞
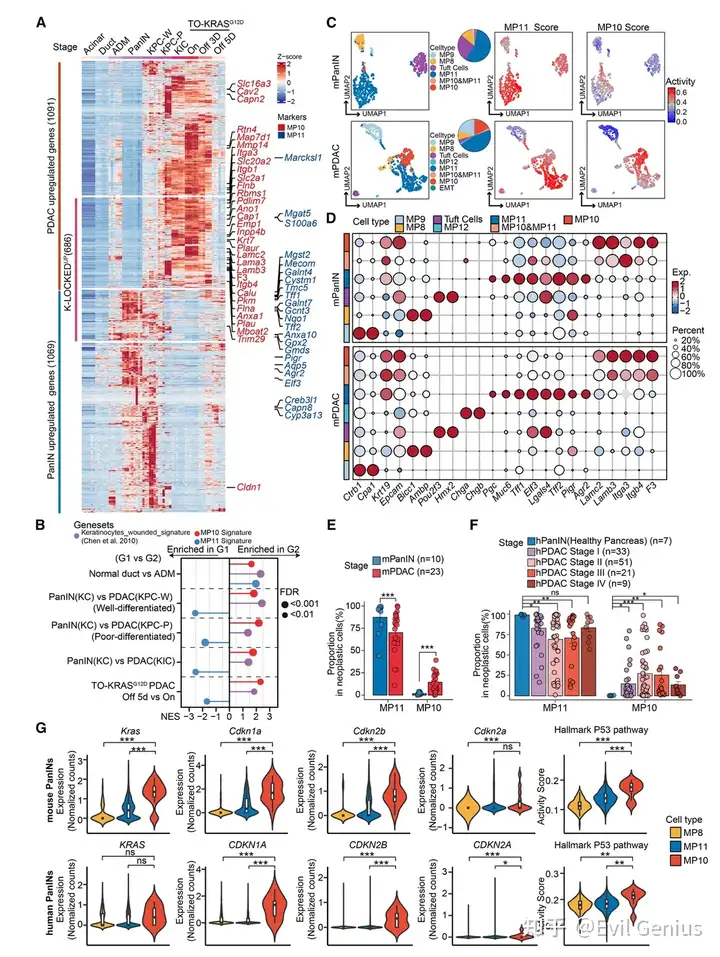
结果2、在小鼠PanIN到PDAC进展过程中,MP10被激活。
小鼠模型中PanIN→PDAC的转录动态
阶段 | 模型 | MP10表达 | MP11表达 |
|---|---|---|---|
正常胰腺 | — | 无 | 无 |
ADM(早期化生) | KC + CAE(2天) | 激活 | 激活 |
PanIN(良性) | KC + CAE(6周) | 关闭/极低 | 进一步激活 |
PDAC(恶性) | KIC(Cdkn2a缺失)或KPC(Trp53缺失) | 强激活 | 减弱 |
关键转变:从PanIN到PDAC,MP11被MP10取代。
KRAS活性对MP10/11的调控
KRASG12D持续激活 → MP10高表达,MP11低表达
KRASG12D耗竭(dox撤除) → MP10下降,MP11上升(时间依赖性)
结论:MP10的维持依赖于高水平的KRAS信号。
单细胞测序揭示的罕见MP10⁺细胞
PanIN中:MP11⁺占主导(>96%),仅存在极少数(<4%)MP10⁺细胞,有时与MP11共表达。
早期浸润性PDAC(TSG缺失后):MP10⁺细胞比例扩大至近30%。
公共数据集验证:人/小鼠PanIN→PDAC均呈现相同趋势。
PanIN中罕见的MP10⁺细胞具有“双面”特征
高表达KRASG12D (驱动癌基因信号)
同时高表达TSG:CDKN1A、CDKN2A、CDKN2B
p53依赖性基因上调
生物学意义:PanIN可自发产生少数试图“恶性转化”的细胞(激活MP10),但同时内源性TSG程序被激活,限制其进一步侵袭——这是一种内在的“刹车”机制。
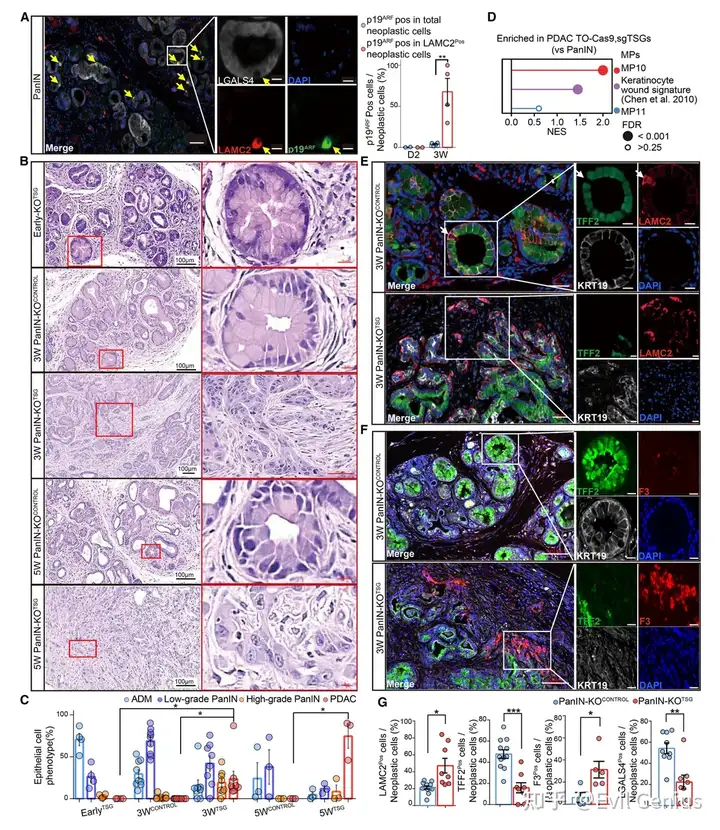
结果3、TSG敲除后PanIN中表达MP10的浸润性细胞快速扩增
PanIN中天然存在罕见的MP10⁺细胞
模型 | 发现 |
|---|---|
人PDAC旁的LG-PanIN | 存在少量MP10表达细胞 |
KC小鼠的LG-PanIN | 存在罕见的、散在的LAMC2⁺/F3⁺/ITGA3⁺细胞 |
这些细胞的特征 | 高pERK(KRAS信号激活)、高表达p19^ARF(Cdkn2a产物) |
即使在没有浸润性癌的良性PanIN中,也存在少数自发激活MP10的细胞,但这些细胞同时高表达TSG蛋白(p19ARF、p16INK4A)。
TSG缺失“释放”MP10细胞,驱动快速恶性转化
实验模型:KC; TO-Cas9(dox诱导Cas9)+ AAV-sgRNA(敲除Cdkn2a、Trp53、Smad4)
分组 | 处理 | 结果(3周后) |
|---|---|---|
KO^CONTROL(对照) | dox 3周 | 仍为LG-PanIN,无浸润 |
PanIN-KO^TSG | dox 3周 | 显著恶性转化:细胞异型性、腺体破坏、小叶结构丧失、浸润 |
TSG缺失后MP10基因表达快速上调(3周内)
RNA-seq证实:TSG敲除后,MP10相关基因表达显著增加
IF证实:浸润细胞高表达MP10标记物(LAMC2、F3、ITGA3),同时下调MP11标记物(TFF2、LGALS4)
PanIN中天然存在一小群“潜伏”的MP10⁺细胞,它们同时被TSG程序(如Cdkn2a、Trp53)所抑制,无法扩增。只有通过TSG缺失解除这种抑制,这些细胞才能快速扩增、关闭MP11、激活MP10,并驱动从良性PanIN向浸润性PDAC的恶性转化。
刹车:TSG(Cdkn2a、Trp53等)→ 抑制MP10⁺细胞的扩增 油门:KRAS突变 → 驱动MP10激活 突破:TSG丢失 → MP10⁺细胞扩增 → 浸润性癌
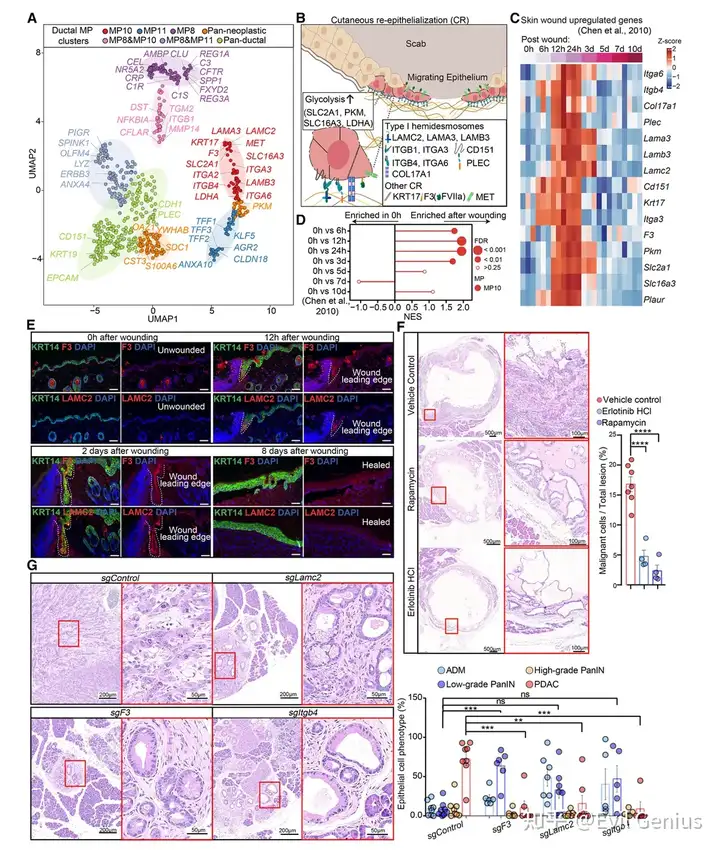
结果4、核心MP10基因重现了皮肤再上皮化(CR)程序
MP10中最特异的基因并非随机分布,而是形成一个功能连贯的模块,与伤口边缘角质形成细胞迁移所需的再上皮化程序完全一致。
MP10特异性基因与皮肤再上皮化(CR)高度重叠
类别 | 具体基因 | 功能 |
|---|---|---|
1型半桥粒组分 | LAMC2, LAMA3, LAMB3, ITGB4, ITGA6, PLEC, CD151, COL17A1 | 基底膜附着、细胞迁移 |
整合素 | ITGA3, ITGB1 | 细胞粘附、迁移 |
角蛋白 | KRT17 | 细胞骨架重塑 |
纤溶酶原激活系统 | F3, PLAU, PLAUR | 基质降解、细胞迁移 |
糖酵解相关 | SLC2A1, SLC16A3, LDHA, PKM | 代谢重编程(Warburg效应) |
MP10基因在皮肤伤口愈合中被动态诱导
小鼠皮肤伤口后12-24小时,MP10基因在迁移的角质形成细胞中达到表达高峰
IF验证:F3和LAMC2在伤口边缘角质形成细胞中特异性诱导,持续至再上皮化完成
靶向CR通路的药物可抑制PDAC侵袭
mTOR抑制剂(雷帕霉素) 或 EGFR抑制剂(厄洛替尼) 处理1周
结果:Matrigel栓块周围的浸润性癌细胞数量显著减少
提示:皮肤伤口愈合所需的信号通路(mTOR、EGFR)同样驱动PDAC的侵袭行为
CR基因是PDAC恶性转化所必需的(体内基因敲除实验)
基因型 | TSG状态 | CR基因状态 | 结果(7周) |
|---|---|---|---|
KC; Cas9 | sgTSG敲除(对照) | 野生型 | 全部出现肉眼可见肿瘤 |
KC; Cas9 | sgTSG敲除 | Lamc2敲除 | 几乎无肿瘤 |
KC; Cas9 | sgTSG敲除 | Itgb4敲除 | 几乎无肿瘤 |
KC; Cas9 | sgTSG敲除 | F3敲除 | 几乎无肿瘤 |
敲除单个CR基因后,其他非靶向的MP10基因表达也随之丢失 → 提示存在正反馈环路
敲除CR基因不影响 ADM形成或ADM→PanIN转变(即早期步骤不依赖CR),但阻断了PanIN→PDAC的恶性进展
MP10核心基因本质上是一个“劫持”了皮肤伤口愈合(再上皮化)程序的转录模块。这些基因(如LAMC2、ITGB4、F3)不仅是PDAC细胞脱离基底膜、浸润实质所必需的,而且它们之间通过正反馈相互维持。靶向这一程序的信号通路(mTOR、EGFR)或直接敲除CR基因,均可有效阻断PDAC的恶性侵袭。
皮肤伤口愈合(生理性) | PDAC恶性进展(病理性) |
|---|---|
│▼伤口边缘角质形成细胞 | │▼PanIN中罕见的MP10⁺细胞 |
│▼再上皮化程序(CR) | │▼劫持 → 同一套CR程序 |
│▼LAMC2, ITGB4, F3, ... | │▼LAMC2, ITGB4, F3, ... |
│▼迁移、覆盖伤口床 | │▼突破基底膜、浸润实质 |
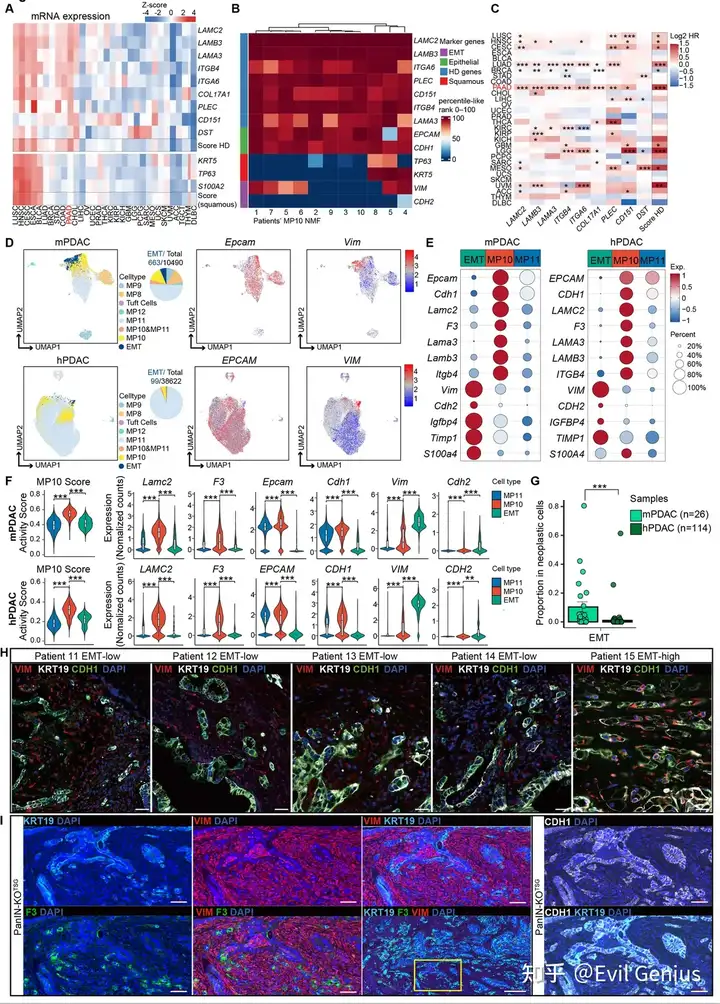
结果5、MP10与鳞状分化及EMT截然不同
MP10 vs 鳞状分化
特征 | 鳞状分化 | MP10/PDAC |
|---|---|---|
1型半桥粒组分 | 高表达 | 高表达 |
鳞状标记(KRT5, TP63) | 高表达 | 低表达 |
谱系限制性 | 广泛 | 限于PDAC、肺癌等特定谱系 |
预后预测价值 | — | HD高表达预示PDAC/肺癌预后差,但对多数癌种无预测价值 |
MP10虽然表达半桥粒等CR基因,但不伴随典型的鳞状分化程序,两者在转录调控上是解偶联的。
MP10 vs EMT
特征 | EMT | MP10 |
|---|---|---|
CDH1/E-cadherin | 抑制 | 高表达(保留) |
EPCAM | 抑制 | 高表达(保留) |
VIM | 诱导 | 几乎不表达(仅极少数部分EMT) |
CDH2/N-cadherin | 诱导 | 不表达 |
细胞形态 | 梭形、失去极性 | 上皮样、保持极性 |
转录状态 | 与MP10 互斥 | 完全EMT细胞中MP10关闭 |
MP10的NMF中:VIM几乎不富集,CDH1和EPCAM强烈富集
单细胞数据:mPDAC中存在EMT细胞群,但MP10⁺细胞与完全EMT细胞互斥
人类PDAC中EMT极其罕见:仅占恶性细胞~0.25%,114个肿瘤中仅5个含>2% EMT细胞
IF验证:11个hPDAC中10个为CDH1⁺/VIM⁻上皮簇,仅1例出现广泛EMT
小鼠 vs 人类
特征 | 小鼠PDAC | 人类PDAC |
|---|---|---|
EMT细胞丰度 | 相对较多(可见EMT细胞簇) | 极其罕见(~0.25%) |
早期浸润中的EMT | 几乎完全不存在 | — |
主要浸润驱动程序 | MP10 | MP10 |
尽管小鼠模型中可见一定程度的EMT,但在早期浸润和人类PDAC中,MP10是主导的、保守的侵袭驱动程序,EMT并非主要机制。
整体比较
MP10程序 | 鳞状分化 | EMT | |
|---|---|---|---|
半桥粒 | +++ | +++ | — |
KRT5/TP63 | — | +++ | — |
E-cadherin | +++ | + | — |
Vimentin | — | — | +++ |
细胞形态 | 上皮样 | 上皮样/分化 | 梭形/间充质 |
在PDAC中 | 主导侵袭 | 罕见 | 极罕见(<0.25%) |
MP10是一个谱系限制性的上皮迁移程序,与鳞状分化无关,与EMT在转录状态上互斥。在PanIN向PDAC的恶性进展中,MP10(而非EMT)是驱动基底膜突破和实质浸润的主导机制。EMT在人类PDAC中极为罕见,且通常发生于晚期肿瘤。
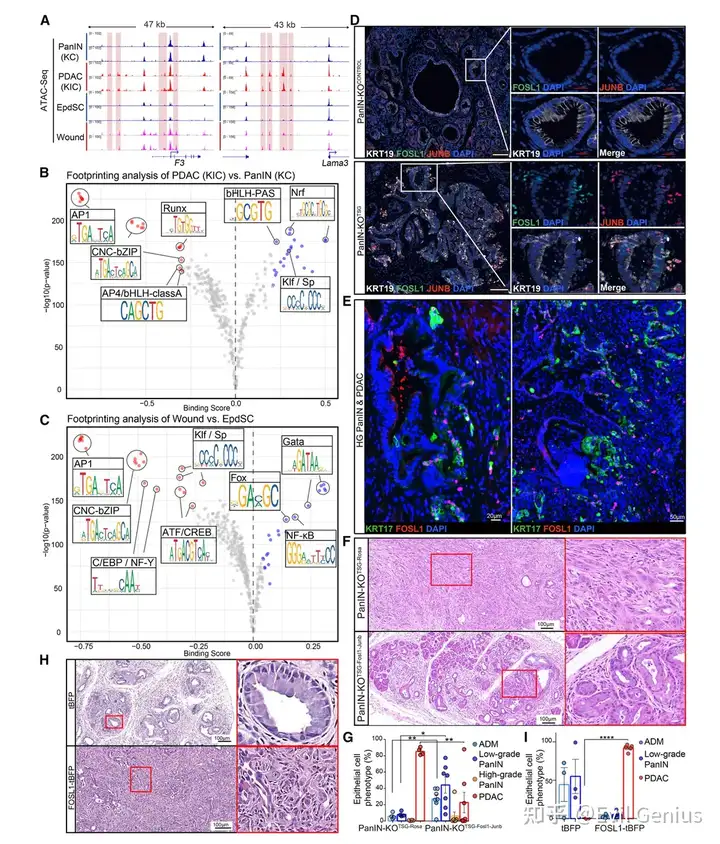
结果6、FOSL1——CR的驱动因子,是PDAC恶性的主调控因子
ATAC-seq
染色质重塑揭示CR与PDAC进展的共享机制
比较组 | 染色质开放位点变化 |
|---|---|
PDAC vs PanIN | >34,000个位点开放增加 |
受伤 vs 未受伤表皮干细胞(CR) | 近一半的共同开放位点 |
共同富集区域 | CR基因位点 |
PDAC恶性进展与皮肤伤口愈合(CR)在染色质水平上高度相似。
AP-1(尤其是FOSL1)是核心转录调控因子
AP-1基序在PDAC vs PanIN和伤口愈合中均为保护程度最强的TF基序
在AP-1家族中,FOSL1是差异最大、升高最显著的成员
必需性筛选:仅有 FOSL1、JUNB、ATF4 对PDAC细胞生长/生存是必需的
谱系限制性:FOSL1在半桥粒高表达肿瘤(PDAC、SCC)中升高
预后价值:在所有AP-1成员中,高FOSL1对hPDAC不良预后预测能力最强
FOSL1/JUNB在空间上定位于侵袭前沿
模型 | 定位 |
|---|---|
皮肤伤口愈合 | 迁移性前沿角质形成细胞核中 |
小鼠PDAC(TSG KO后3周) | 新生浸润性细胞核中(特异性共表达) |
人类PDAC | 从导管新生的MP10⁺浸润性细胞核中 |
FOSL1的表达受KRAS信号及CR通路药物调控
KRASG12D 耗竭 → FOSL1蛋白 ↓、MP10基因 ↓
雷帕霉素(mTOR抑制剂)或厄洛替尼(EGFR抑制剂)→ FOSL1⁺浸润性细胞 ↓
遗传学验证:FOSL1/JUNB是PDAC恶性转化的必要条件
实验 | 结果 |
|---|---|
TSG KO(对照组) | 快速出现浸润性PDAC |
TSG KO + Fosl1/Junb共敲除 | 浸润性PDAC显著减少 |
FOSL1和JUNB是TSG缺失后恶性转化所必需的。
FOSL1单独足以驱动全胰腺恶性转化(无需TSG缺失)
实验组 | 结果(3周后) |
|---|---|
KC + tBFP(对照) | LG-PanIN,无浸润 |
KC + FOSL1-tBFP | 全胰腺恶性转化:小叶结构丧失、高度非典型细胞全胰腺迁移 |
FOSL1过表达不依赖TSG缺失即可驱动浸润性癌
即使在p19ARF广泛激活(TSG程序被激活)的情况下,FOSL1仍能驱动恶性表型
这一发现极为重要:FOSL1的强制表达可绕过TSG介导的肿瘤抑制屏障,直接将良性PanIN转化为浸润性癌。
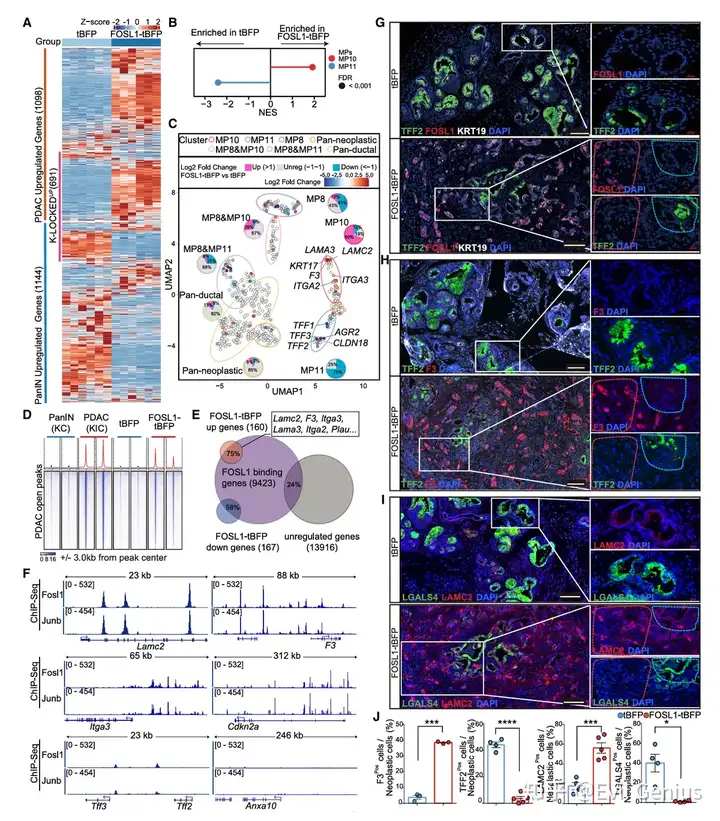
结果7、FOSL1结合并激活MP10 CR基因
FOSL1过表达的表型与自然进展高度一致
对比项 | FOSL1过表达 vs 自然进展(PanIN→PDAC) |
|---|---|
转录变化 | 高度镜像 |
上调基因 | 富集 MP10基因(包括几乎所有CR基因) |
下调基因 | 富集 MP11基因 |
染色质开放 | 与TSG KO诱导的进展高度相似 |
上皮/间充质状态 | 高Epcam/Cdh1,低Vim(上皮状态,非EMT) |
FOSL1单因子足以重演PanIN→PDAC进展过程中的核心转录和表观遗传变化。
FOSL1的调控具有选择性
基因类别 | 受FOSL1影响? |
|---|---|
多个MP共有的泛导管基因(如KRT19, EPCAM) | 基本不受影响 |
MP10特异性基因(尤其是CR基因) | 选择性激活(几乎所有) |
MP11特异性基因 | 抑制 |
FOSL1并非广谱转录激活因子,而是精准调控MP10/MP11转换的关键开关。
FOSL1直接结合并调控靶基因(ChIP-seq证据)
指标 | 数值 |
|---|---|
FOSL1与JUNB结合位点的重叠度 | 非常高(符合异二聚体功能) |
上调基因中FOSL1结合的比例 | ~75% |
下调基因中FOSL1结合的比例 | ~58% |
非调控基因中FOSL1结合的比例 | <25% |
激活基因上的结合强度 | 高于抑制基因 |
直接靶向的CR基因示例:LAMC2、F3等恶性转化必需基因
FOSL1同时激活致癌程序和TSG程序
FOSL1-tBFP表达 → Cdkn2a mRNA ↑、p19^ARF蛋白 ↑
ChIP-seq证实:FOSL1/JUNB直接结合Cdkn2a的长距离增强子(Ncruc)
该增强子在自发性PDAC中频繁缺失(说明肿瘤通过删除此增强子来逃避FOSL1介导的TSG激活)
重要机制:FOSL1同时激活:
MP10 CR基因(驱动恶性)
Cdkn2a/TSG程序(限制恶性)
这解释了为什么在自然进展中需要额外的TSG缺失——FOSL1自带的“刹车”需要被解除才能实现完全恶性转化。
LAMC2是FOSL1驱动恶性所必需的
实验 | 结果 |
|---|---|
FOSL1-tBFP(对照) | 侵袭性表型(细胞簇从腺体出芽) |
FOSL1-tBFP + Lamc2 KO | 侵袭表型完全逆转,FOSL1表达细胞局限于PanIN样结构 |
结论:LAMC2是FOSL1下游的关键效应分子,对于FOSL1驱动的恶性浸润是不可或缺的。
蛋白水平验证(IF)
FOSL1诱导的肿瘤中 | 表达变化 |
|---|---|
MP10 CR因子(LAMC2、F3) | 激活 |
MP11标记物(LGALS4、TFF2) | 下调 |
细胞形态 | 形成小簇,从残余腺体“出芽” |
FOSL1是直接结合并激活MP10 CR基因的主转录因子。它选择性地上调MP10程序(包括LAMC2、F3等关键侵袭基因)并抑制MP11程序,同时意外地也直接激活Cdkn2a(TSG)增强子,形成一种“双刃剑”效应:驱动恶性的同时内在地激活了限制机制。这解释了为何FOSL1单独足以启动恶性转化,但完全进展需要额外的TSG缺失。LAMC2是FOSL1下游的关键效应分子,敲除它可完全逆转FOSL1诱导的侵袭表型。
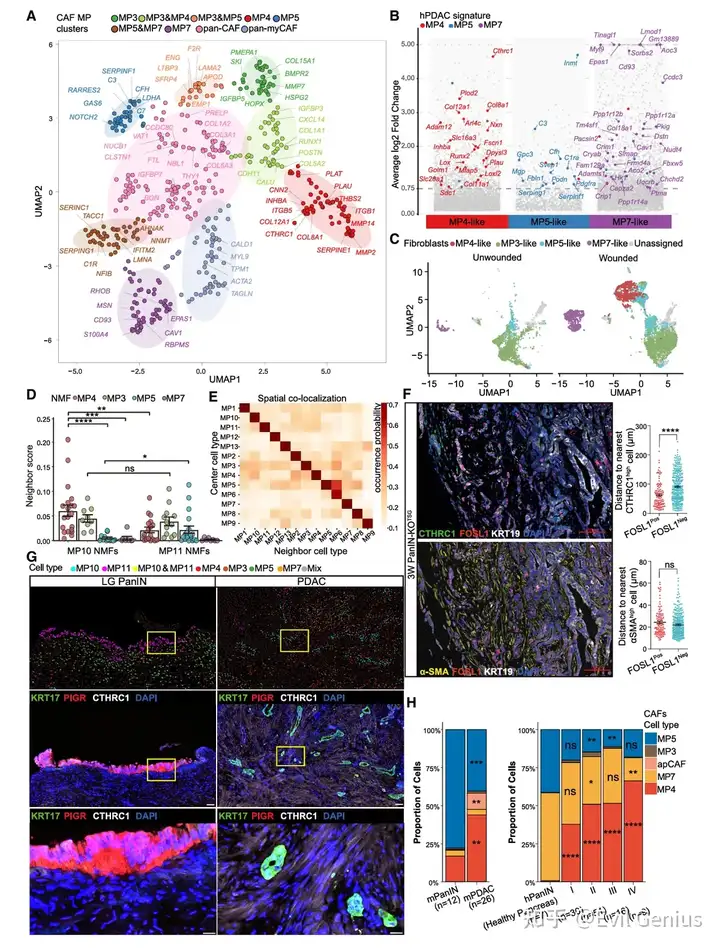
结果8、CTHRC1高表达的MP4 myCAF特异性结合恶性肿瘤细胞
肿瘤微环境影响癌细胞转录状态
成纤维细胞MP的分类与特征
MP | CAF类型 | 特异性标记物 | 共享标记物 |
|---|---|---|---|
MP4 | myCAF亚型 | CTHRC1, COL12A1 | ACTA2, MYL9, TAGLN |
MP3 | myCAF亚型 | IGFBP5, HOPX | ACTA2, MYL9, TAGLN |
MP7 | myCAF亚型 | EPAS1 | ACTA2, MYL9, TAGLN |
MP5 | iCAF | 补体基因等 | — |
CAF程序与皮肤伤口愈合的对应关系
CAF MP | 是否在未受伤皮肤成纤维细胞中表达 | 是否在受伤皮肤成纤维细胞中表达 |
|---|---|---|
MP3 | ✓ | ✓ |
MP5 | ✓ | ✓ |
MP7 | ✓ | ✓ |
MP4 | ✗ | ✓(仅伤口特异性) |
MP4特征仅在伤口特异性成纤维细胞中表达,提示PDAC中的MP4 CAF相当于肿瘤中的“伤口相关成纤维细胞”。
不同CAF MP与肿瘤细胞的空间邻近关系
CAF MP | 与MP10(恶性)的邻近性 | 与MP11(良性)的邻近性 |
|---|---|---|
MP7 | 几乎不邻近(任何肿瘤细胞) | 几乎不邻近 |
MP5(iCAF) | 不邻近 | 偶尔邻近 |
MP3 | 相似 | 相似 |
MP4(CTHRC1高) | 显著富集(最强、最特异) | 较弱 |
CTHRC1高表达的MP4 myCAF特异性定位在恶性MP10⁺肿瘤细胞周围,而其他CAF类型则无此特异性。
从PanIN到PDAC的CAF动态变化
CAF MP | 在PanIN中的丰度 | 在PDAC中的丰度 | 趋势 |
|---|---|---|---|
MP5(iCAF) | 丰富 | 减少 | 随进展下降 |
MP4(CTHRC1高) | 稀少 | 增加 | 随进展上升(与MP10⁺细胞扩增同步) |
随着PanIN→PDAC进展,CAF群体从MP5(iCAF)主导转向MP4(CTHRC1高myCAF)主导,与肿瘤细胞中MP10的激活和扩增在时空上同步。
蛋白水平验证(IF)
CTHRC1(而非α-SMA)的表达可预测CAF与FOSL1⁺浸润性PDAC细胞的邻近关系
在小鼠和人类PDAC中均得到验证
总结一下啊
PDAC中存在四种功能不同的成纤维细胞MP。其中,MP4(CTHRC1高表达的myCAF亚型)具有以下特征:
伤口特异性:仅在皮肤伤口愈合中的成纤维细胞中表达,提示其代表一种“伤口相关”CAF状态;
空间特异性:选择性定位在恶性MP10⁺肿瘤细胞周围,而不靠近良性MP11⁺细胞;
动态特异性:在PanIN→PDAC进展过程中数量增加,与MP10⁺恶性细胞扩增同步;
分子标记物:CTHRC1是比α-SMA更精确的恶性相关CAF标记物。
这些发现强烈提示,MP4 CAF与MP10⁺恶性肿瘤细胞之间存在功能性的相互作用,共同驱动PDAC的恶性进展。
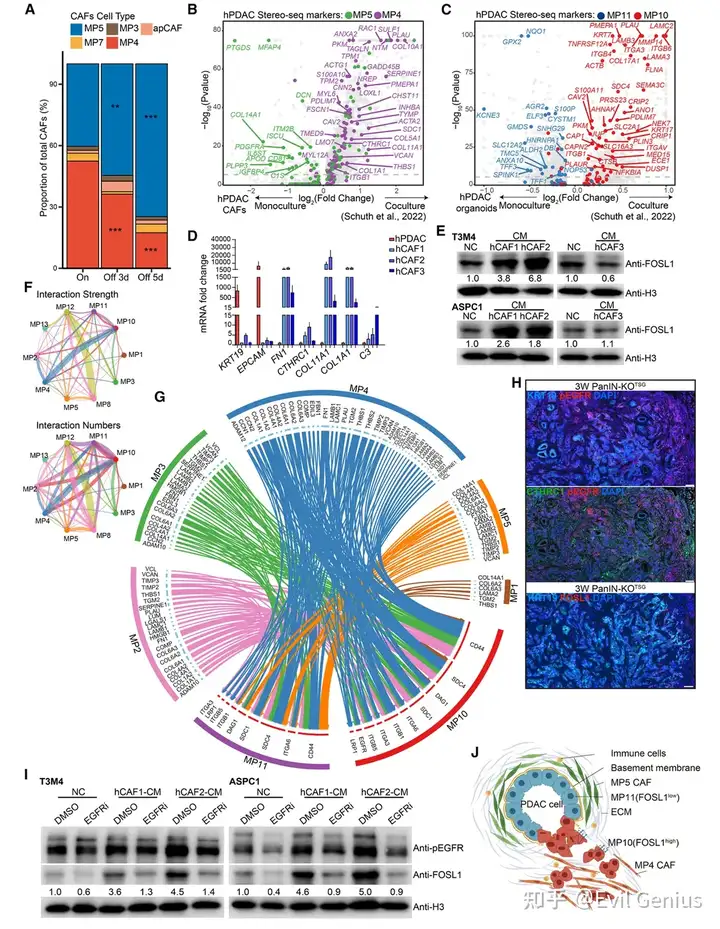
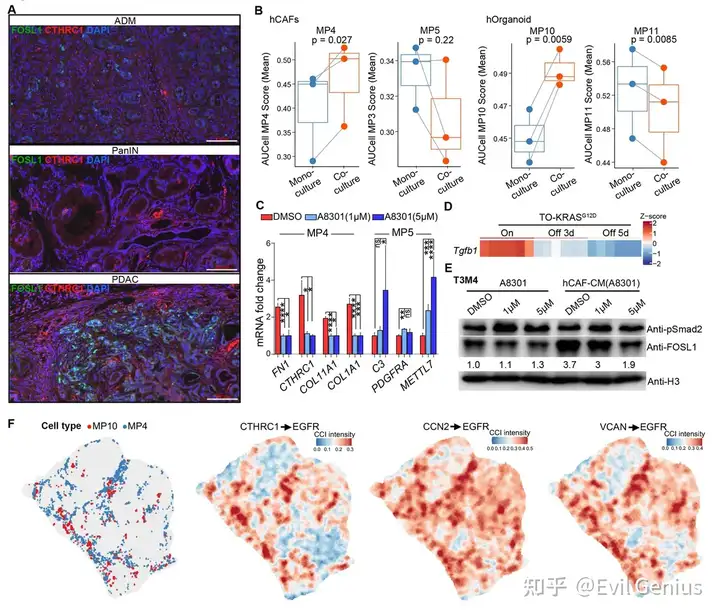
结果9、MP4样CAF通过肿瘤细胞EGFR激活促进FOSL1表达
MP4与MP10在功能上相互依赖
实验 | 结果 |
|---|---|
KRASG12D耗竭(dox撤除) | MP4 CAF比例↓,MP5 iCAF成为主导 |
hPDAC类器官 + CAF共培养 | CAF中MP4基因↑;PDAC类器官中MP10基因↑ |
MP10⁺肿瘤细胞与MP4 CAF之间存在双向互作,两者互相维持。
MP4样CAF特异性诱导PDAC细胞中FOSL1表达
CAF来源 | 条件培养基效果 |
|---|---|
MP4样CAF(2个细胞系) | FOSL1水平显著升高 |
MP5样CAF(1个细胞系) | 无显著效果 |
是MP4 CAF的分泌因子(而非MP5 CAF)特异性上调FOSL1。
MP4 CAF状态受TGF-β信号调控
处理 | 效果 |
|---|---|
TGF-β抑制剂(A8301)处理MP4 CAF | MP4基因↓,MP5基因↑ |
该处理CAF的条件培养基 | 激活FOSL1能力减弱 |
直接加抑制剂到未条件化培养基 | 不影响基础FOSL1或p-SMAD2 |
TGF-β信号维持MP4 CAF状态;KRAS突变通过上调Tgfb1间接调控CAF状态。
空间细胞通讯分析:MP4 → MP10是最强信号轴之一
基于Stereo-seq的空间邻近性 + 基因表达预测细胞间通讯
MP4-MP10相互作用被评为最强检测到的相互作用之一
MP4 CAF通过非经典配体激活肿瘤细胞EGFR
MP4CAF表达的因子 | 类型 | 已知功能 |
|---|---|---|
CCN2 | 非经典EGFR激活因子 | 激活EGFR |
VCAN | 非经典EGFR激活因子 | 激活EGFR |
CTHRC1 | 非经典EGFR激活因子 | 激活EGFR(且为MP4特异性标记) |
经典EGFR配体(如EGF) | 有限富集 | — |
实验验证:EGFR是MP4→FOSL1信号通路的关键节点
实验 | 结果 |
|---|---|
MP4样CAF条件培养基 | hPDAC细胞中p-EGFR(Y1068)↑ |
+ EGFR抑制剂厄洛替尼 | 完全阻断FOSL1诱导效应 |
IF共定位 | FOSL1⁺浸润性PDAC区域中,EGFR激活与CTHRC1⁺ CAF空间邻近 |
MP4 CAF与MP10⁺ PDAC细胞之间形成一个双向互作的恶性环路:
MP4 CAF通过分泌非经典EGFR配体(CTHRC1、CCN2、VCAN等)激活肿瘤细胞EGFR信号;
EGFR激活 → 肿瘤细胞中FOSL1表达上调 → FOSL1驱动MP10 CR基因表达 → 肿瘤细胞获得侵袭能力;
同时,MP10⁺肿瘤细胞通过KRAS依赖的方式(如TGF-β)维持MP4 CAF状态;
这一互作环路解释了MP4与MP10在空间上的高度邻近性和时间上的同步扩增。
靶向EGFR(厄洛替尼)可阻断MP4 CAF对FOSL1的诱导效应,为PDAC治疗提供了理论依据。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号