分子动力学--非标残基的处理五(配体)
原创
分子动力学--非标残基的处理五(配体)
原创
追风少年i
发布于 2026-05-10 09:01:21
发布于 2026-05-10 09:01:21
作者,Evil Genius
前面一直分析,想要采用Gaussian一步计算完优化 + 频率 + 生成gesp文件,事实证明不可取,必须拆分两步。
不能一步获取的原因也很简单
Pop=MK + IOp(6/50=1) → 强制导出静电势 / 电荷 SMD + Water → 连续介质模型,L602 要处理溶剂场 Br 原子(重卤素) → 半径、网格、势拟合非常敏感 opt + freq + Pop 一次性完成 → L602 在复杂波函 + 溶剂下极易越界 %mem=24GB / %nprocshared=24 → 放大并发内存访问 → 更容易触发段错误 ✅ 这是 Gaussian 官方邮件列表里反复出现的“已知不稳定组合”
最佳最好还是要拆分成2步
第一步:只优化 + 频率
配置文件
%chk=ligand.chk
%nprocshared=16
%mem=16GB
#P B3LYP/6-31G(d) opt freq SCRF=(SMD,Solvent=Water)
Title: ligand.pdb
0 1
O 4.68725 -0.14473 4.41003
O 7.53782 0.25274 3.90488
N 3.21339 -2.51671 0.04448
N 7.35642 -2.47520 0.13790
N 5.30833 -3.09637 -0.90452
C 5.28155 -1.97812 1.22860
C 6.67341 -1.92346 1.14695
C 5.33471 -0.73999 3.35912
C 4.63052 -1.41572 2.35526
C 6.71662 -0.58837 3.19913
C 4.63076 -2.54918 0.11926
C 7.36022 -1.21004 2.11967
C 2.44552 -2.40899 -1.13628
C 3.52868 -0.83399 4.91033
C 6.63667 -3.04949 -0.83409
C 7.51333 0.16225 5.32515
C 1.12841 -2.88283 -1.16702
C 2.92339 -1.71999 -2.26438
C 2.23305 -0.33719 4.27481
C 6.94948 1.44376 5.91664
C 0.34757 -2.72586 -2.31674
C 2.14260 -1.55949 -3.41054
C 0.85045 -2.07070 -3.43948
H 3.54909 -1.46484 2.38248
H 8.43793 -1.09716 2.01506
H 2.81343 -3.13219 0.74253
H 3.48426 -0.63904 5.98796
H 3.63349 -1.92255 4.80820
H 8.55876 0.06671 5.64818
H 6.99215 -0.72947 5.68783
H 7.18787 -3.50773 -1.64889
H 0.70716 -3.38286 -0.30009
H 3.92519 -1.28955 -2.25468
H 2.40576 0.33902 3.43243
H 1.63413 -1.18660 3.92448
H 1.61844 0.18984 5.01146
H 6.30780 1.97831 5.20995
H 6.38464 1.22724 6.82745
H 7.76178 2.12509 6.19638
H 2.54636 -1.03071 -4.26979
H 0.24578 -1.94587 -4.33320其中复习一下参数的意义
参数 | 含义 | 作用 |
|---|---|---|
#P | 详细输出模式 | Print 的缩写,让输出文件包含更详细的信息,便于调试和分析 |
B3LYP | 密度泛函方法 | Becke 三参数杂化泛函 + Lee-Yang-Parr 相关泛函,是目前有机化学中最流行的 DFT 方法之一 |
6-31G(d) | 基组 | 6-31G 加极化函数,对非氢原子添加 d 轨道,是 DFT 计算的标配基组 |
opt | 几何优化 | 寻找分子的稳定构型(能量极小点) |
freq | 频率分析 | 计算振动频率,用于验证优化点是否为真正的极小点(无虚频),同时可获得热力学量(熵、焓、自由能) |
SCRF | 自洽反应场 | 使用连续溶剂模型,将溶剂效应纳入计算 |
SMD | 溶剂模型 | Solvation Model based on Density,一种通用连续溶剂模型,对水和有机溶剂都有较好表现 |
Solvent=Water | 溶剂类型 | 指定溶剂为水 |
然后我们运行
g16 < ligand.gjf > ligand.out生成chk文件
第二步:单独生成 .gesp(读 chk)
配置文件
%chk=ligand.chk
%nprocshared=8
%mem=16GB
#P B3LYP/6-31G(d) Pop=MK IOp(6/33=2) IOp(6/42=6) IOp(6/50=1) geom=check
ligand
0 1
ligand.gesp然后运行
g16 < ligand.gjf > ligand.out就会生成我们需要的gesp文件,这个过程也比较长。
还有就是电荷模式,我们这里采用的是Pop=MK,除了这个,还有其他,总结如下
电荷模型 | Gaussian 关键词 | 采样/计算方法特点 | 适用场景与备注 |
|---|---|---|---|
Merz-Kollman | Pop=MK | 在多层Connolly分子表面上选取采样点。你正在使用的方法,在Gaussian中应用广泛。 | 对分子构象的依赖性相对较小。遇到的 l602.exe 报错与此方法的高计算需求有关。 |
CHELPG | Pop=CHELPG | 在围绕分子的规则立方体网格上选取采样点。 | 对分子对称性的表现通常更好,电荷在不同构象下更稳定。一个重要的优点是,对于Gaussian没有内置范德华半径的原子(如溴 Br),它不容易崩溃。 |
RESP | Pop=(MK,RESP=N) | 两步拟合 + 施加惩罚函数。首先无约束拟合,然后对非氢原子施加约束(如电荷相等),最后再拟合一次氢原子电荷。在Gaussian 16 Rev. C.01及以上版本中可用。 | 分子动力学模拟(尤其是Amber力场)的黄金标准。通过约束确保了化学等价原子电荷的一致性,对柔性分子尤其重要。 |
CHelp | Pop=CHelp | 一种较老的拟合方案,采样点也基于分子表面。 | 使用较少,现在基本被 CHelpG 和 MK 取代。 |
HLY | Pop=HLY | 另一种早期的ESP拟合方案。 | 现在不常用。 |
最后生成带电荷的mol2文件
antechamber -i ligand.gesp -fi gesp -o ligand.mol2 -fo mol2 -pf y -c resp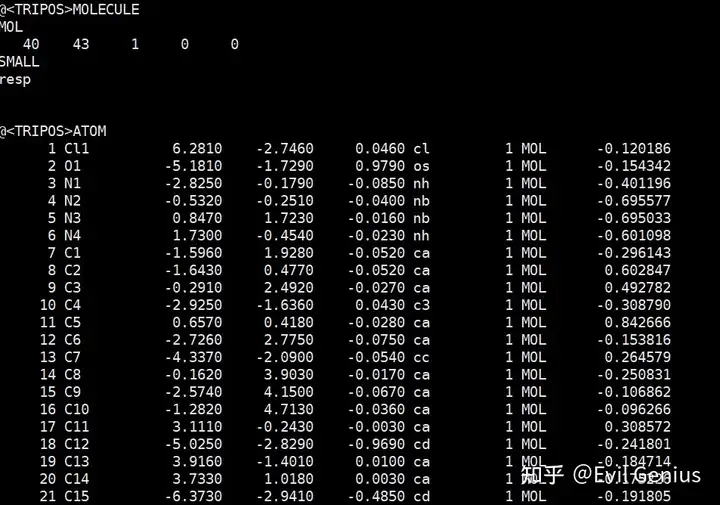
用这个文件进行sobtop是最好。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号