分子动力学--非标残基的处理六(配体)
原创
分子动力学--非标残基的处理六(配体)
原创
追风少年i
发布于 2026-05-11 10:03:40
发布于 2026-05-11 10:03:40
作者,Evil Genius
今天这一篇我们需要用之前Gaussian16 + antechamber 生成的mol2文件生成Gromacs需要的gro文件。
我们用两种方法都做一下
第一种,sobtop
首先要处理文件,到处都是坑,之前生成的是符合Amber/GAFF 原子类型,但是sobtop要求必须是Tripos 格式。
我们来处理一下,先来看一下
GAFF立场格式
@<TRIPOS>MOLECULE
MOL
40 43 1 0 0
SMALL
resp
@<TRIPOS>ATOM
1 Cl1 6.2810 -2.7460 0.0460 cl 1 MOL -0.120186
2 O1 -5.1810 -1.7290 0.9790 os 1 MOL -0.154342
3 N1 -2.8250 -0.1790 -0.0850 nh 1 MOL -0.401196
4 N2 -0.5320 -0.2510 -0.0400 nb 1 MOL -0.695577
5 N3 0.8470 1.7230 -0.0160 nb 1 MOL -0.695033
6 N4 1.7300 -0.4540 -0.0230 nh 1 MOL -0.601098
7 C1 -1.5960 1.9280 -0.0520 ca 1 MOL -0.296143
8 C2 -1.6430 0.4770 -0.0520 ca 1 MOL 0.602847需要转化的Tripos标准格式
@<TRIPOS>MOLECULE
MOL
40 43 1 0 0
SMALL
resp
@<TRIPOS>ATOM
1 Cl1 6.2810 -2.7460 0.0460 Cl 1 MOL -0.120186
2 O1 -5.1810 -1.7290 0.9790 O.2 1 MOL -0.154342
3 N1 -2.8250 -0.1790 -0.0850 N.pl3 1 MOL -0.401196
4 N2 -0.5320 -0.2510 -0.0400 N.ar 1 MOL -0.695577
5 N3 0.8470 1.7230 -0.0160 N.ar 1 MOL -0.695033
6 N4 1.7300 -0.4540 -0.0230 N.pl3 1 MOL -0.601098
7 C1 -1.5960 1.9280 -0.0520 C.ar 1 MOL -0.296143
8 C2 -1.6430 0.4770 -0.0520 C.ar 1 MOL 0.602847
9 C3 -0.2910 2.4920 -0.0270 C.ar 1 MOL 0.492782软件兼容性之间真的还需要小心处理。
第一步:输入mol2文件
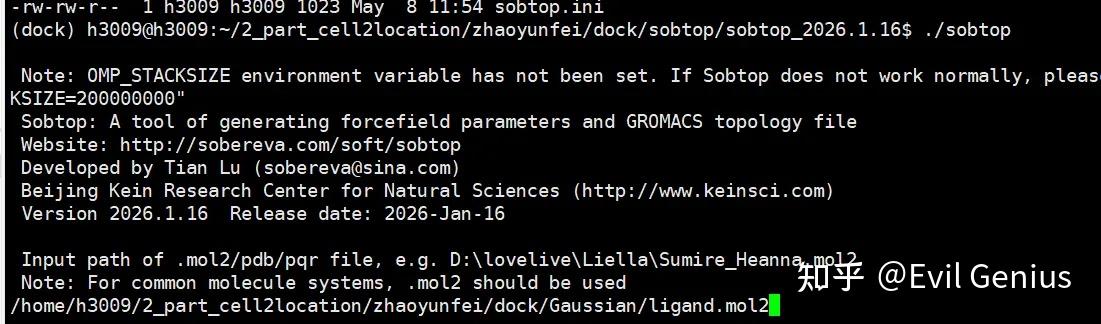
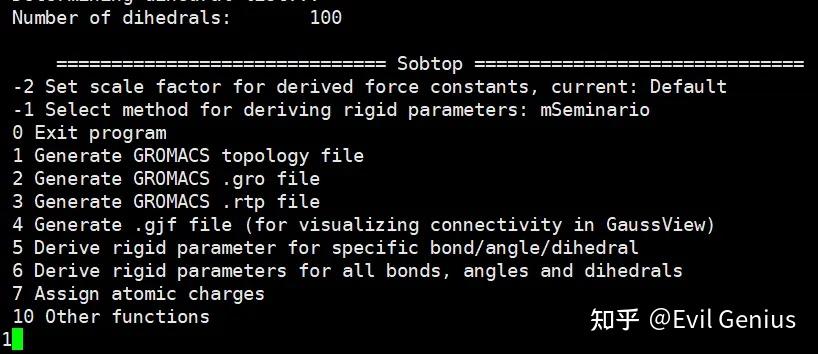
第二步:生成gro文件,选择1
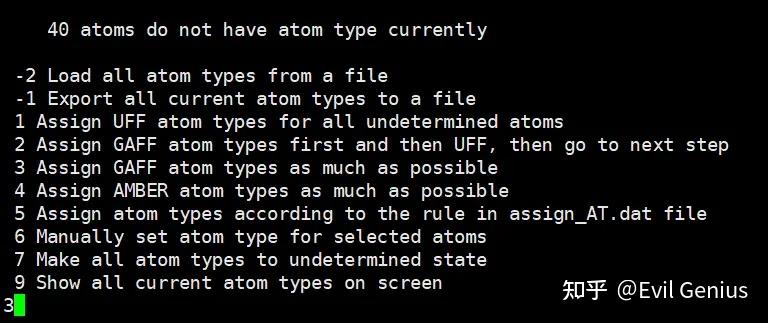
第三步:优先分配 GAFF 类型(做分子动力学(GROMACS/Amber)的标准流程,配合你的高斯 RESP 电荷)
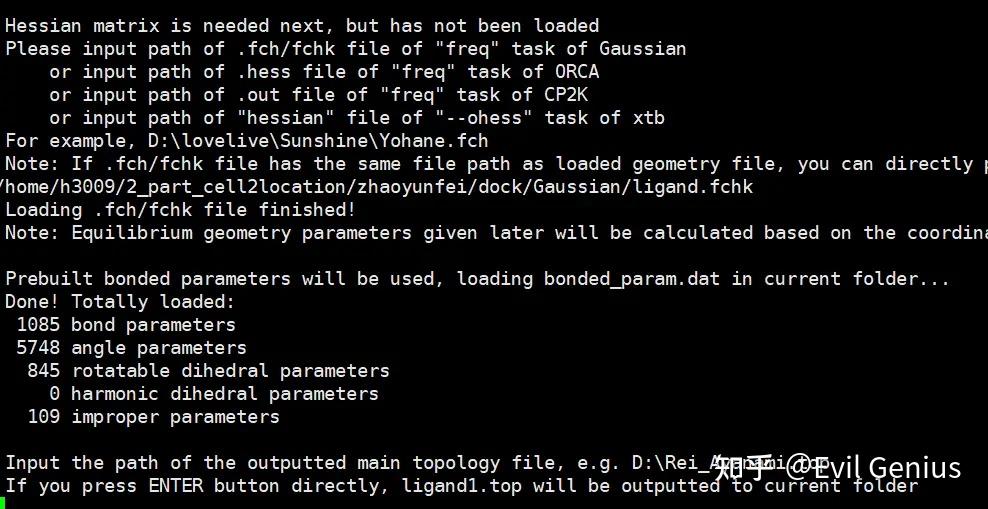
第四步:从 Hessian 矩阵精确推导力常数。
第五步:指定输出目录
这样就生成了我们的gro、itp、top文件。
第二种方法
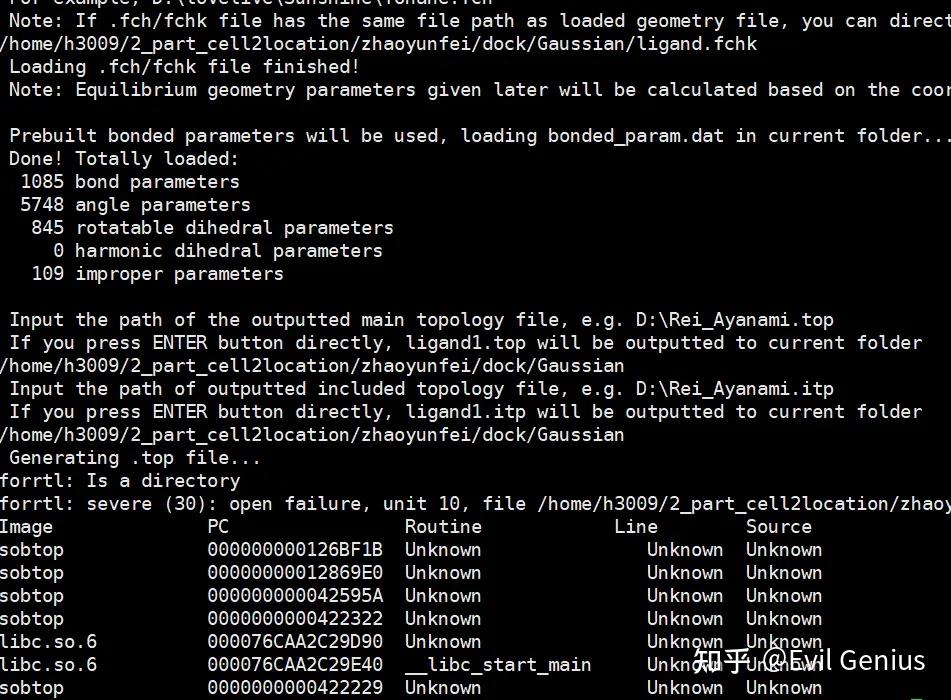
运行,这个时候不需要再转化ligand.mol2的数据格式。
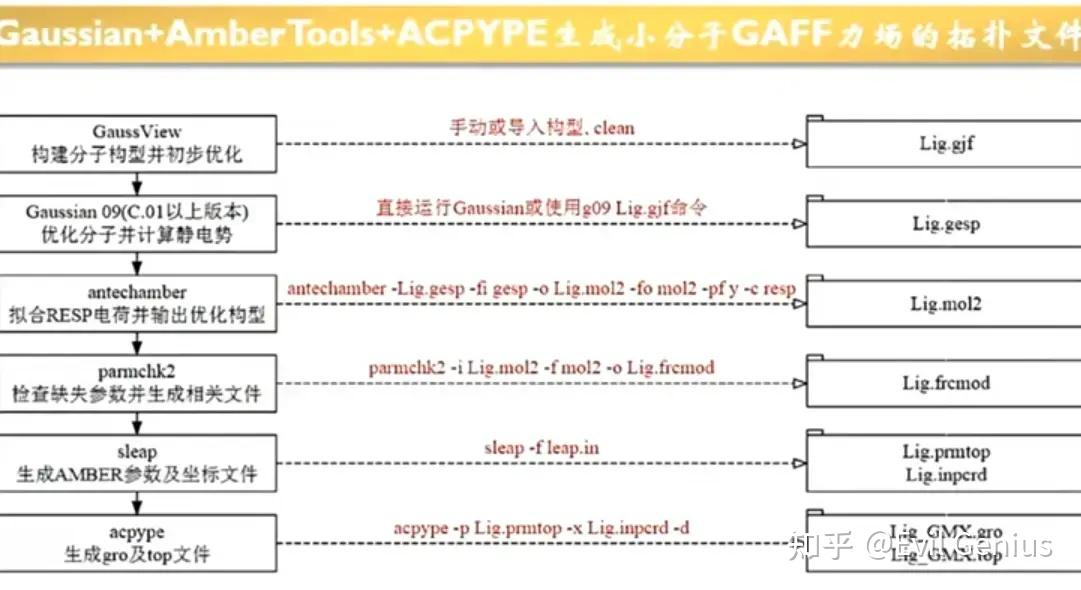
parmchk2 -i ligand.mol2 -f mol2 -o ligand.frcmod创建文件http://tleap.in
# 1. 加载小分子力场 (这是基本参数集)
source leaprc.gaff
# 2. 加载你的配体分子结构
# 这里的 "LIG" 可以自定义,是你给这个分子起的名字
# ligand_tripos.mol2 是你之前准备好的、原子类型正确的 Tripos 格式文件
LIG = loadmol2 ligand.mol2
# 3. 加载你的配体补充参数文件
# 这个命令会读入你上一步生成的 ligand.frcmod 文件,
# 用它来补全上一步加载的 LIG 分子所缺失的参数
loadamberparams ligand.frcmod
# 4. (强烈推荐) 检查分子是否有问题
# 如果输出中有关于原子参数缺失的错误,需要回去检查 frcmod 文件
check LIG
# 5. (可选) 保存为库文件,方便以后重复使用
saveoff LIG ligand.lib
# 6. 输出用于模拟的拓扑和坐标文件
# 这一步会生成你想要的 ligand.prmtop 和 ligand.inpcrd
saveamberparm LIG ligand.prmtop ligand.inpcrd
# 7. 退出程序
quit运行
tleap -f leap.in最后生成gromacs的拓扑文件
acpype -p ligand.prmtop -x ligand.inpcrd -d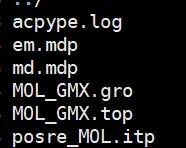
生成了我们需要的文件
哪种更好呢?明显是第二种
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号