分析梳理--分子动力学模拟的常规步骤九(Gromacs)
原创
分析梳理--分子动力学模拟的常规步骤九(Gromacs)
原创
追风少年i
发布于 2026-05-17 08:38:07
发布于 2026-05-17 08:38:07
作者,Evil Genius
这一篇我们继续分子动力学,对蛋白配体复合物的NVT、NPT预平衡。
先来看NPT预平衡
平衡蛋白质-配体复合物与平衡任何其他蛋白质水溶液体系一样.但需要有些特殊考虑:
1、对配体施加限制
2、处理温度耦合组
蛋白文件的限制文件在一开始的pdb2gmx已经施加了限制。
gmx genrestr -f MOL.gro -o posre_MOL.itp -fc 1000 1000 1000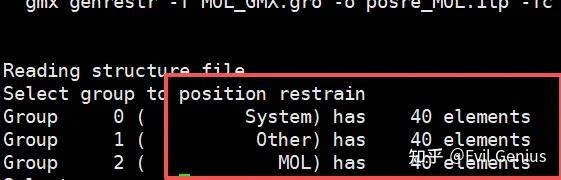
这个时候三个组选择哪个都可以。
生成配体的位置限制性文件posre_MOL.itp
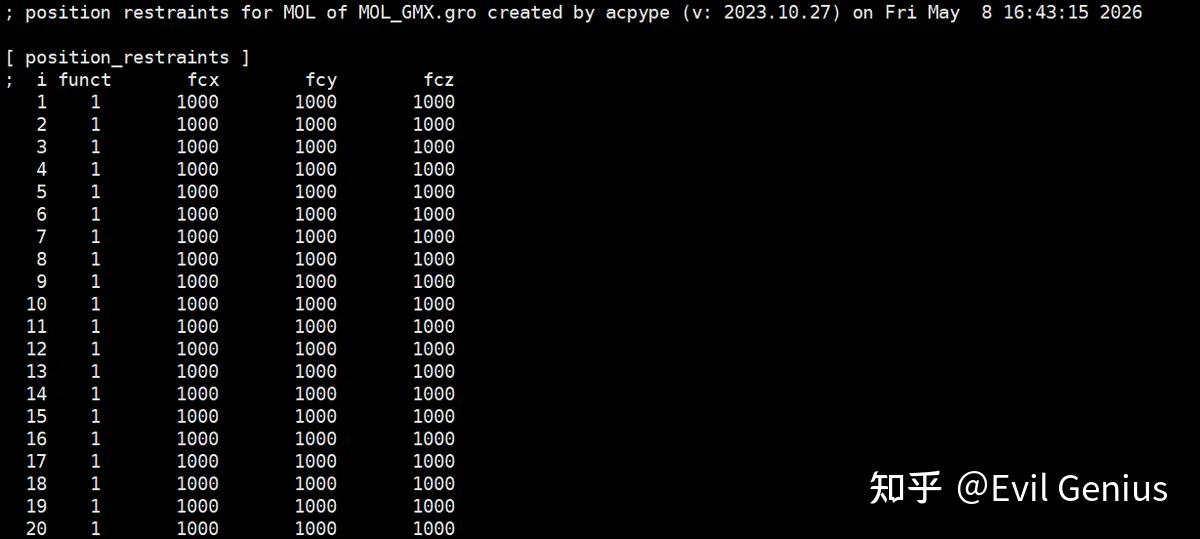
gmx Genrestr根据-f文件的内容为包含原子序数列表和x、y和Z方向的三个力常数的拓扑生成一个#include文件。可以在命令行上给出单个各向同性力常数而不是三个分量。
看一下主要参数
-f[<.gro/.g961...>] (conf.gro)输入配体的结构性文件
-fc <vector>(1000 1000 1000) 力常数(k/mol nm2)
-o[<.itp>] (posre.itp)输出配体的限制性文件
然后需要在拓扑文件中包含配体的位置限制信息。在限制性预平衡中,蛋白质被限制,也要简单地限制配体的位置,将下面几行添加到拓扑文件中的指定位置:

拓展(了解):通常我们不需要单独的设置。
如果在平衡过程中需要更多的控制,想要独立地限制蛋白质和配体,可以使用不同的#ifdef块来控制配体的包含位置限制文件的位置:

热浴耦合组的设置
温度耦合的正确控制是非常敏感的事。将每个分子类型单独耦合到其自身的热浴组是不恰当的,不利于体系的稳定。例k:tc_grps= Protein MOLSOLCL体系可能会奔溃,因为对只有含有几个原子的热浴组(如MOL和CL),在控制其动能的涨落时,温度耦合算法不够稳定。不要对体系中的每一单个类型分子使用独立的耦合。
典型的方法是设定tc_grps= Protein Non-Protein进行升温模拟。 但Non-Protein组也包含配体分子MOL,实际上配体与蛋白质物理上结合非常紧密,最好将它们一起考虑。那么可以在温度耦合时将MOL当作蛋白质的一部分。另外把添加的离子作为溶剂的一部分.进行耦合。
需要一个特殊的分组,其中包含蛋白质和配体分子MOL
gmx make_ndx -f em.gro -o index.ndx生成包含蛋白和配体为整体的索引组index.ndx。
几乎每个GROMACS程序都需要索引组。所有这些程序都可以生成默认索引组。只有在需要特殊索引组时才需要使用gmxmake_ndx。整个系统有一个默认索引组。
使用索引编辑器,可以选择原子、残基和链的名称。和编号。当提供运行输入文件时,还可以选择原子类型。
看一下参数
-f[<.grol.g96/...>] (conf.gro)只要提供完整体系的任意坐标文件即可。在这里,使用em.gro,它是体系(能量最小化后)的输出结构。
-o[<.ndx>] (index.ndx)输出的包含特定组的索引文件
将Protein和MOL组合并在一起,其中的>为提示符:
>1|13 >q
现在可以设置tc_grps= Protein_MOL Water_and_ions来达到需要的Protein Non-Protein的效果了.
我们来运行一下,下面是默认索引组,有Water_and_ions分组,没有Protein_MOL组,需要我们建立。
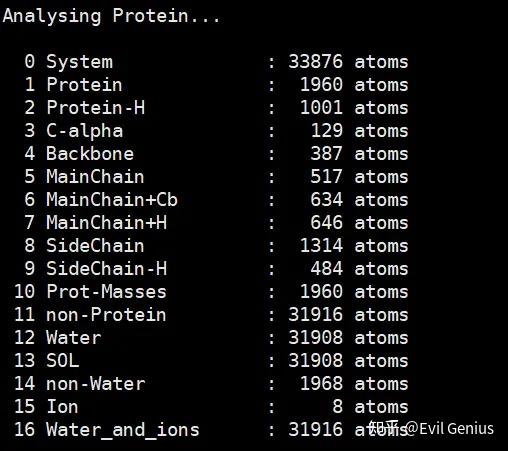
我们输入1|13,然后q保存退出,就新建了Protein_MOL组。
那么正常运行NVT的预平衡就可以了。
gmx grompp -f nvt.mdp -c em.gro -p topol.top -o nvt.tpr -r em.gro -n index.ndx使用GROMACS的grompp(GROMacs Pre-Processor)模块产生nvt.tpr文件,下一步执行NVT预平衡模拟所需的输入文件,后续也要使用这个功能,使用位置约束时,必须使用-r提供具有约束坐标的文件(可以与为-c提供的文件相同)。
gmx mdrun -deffnm nvt使用GROMACS的mdrun模块执行整个体系的NVT预平衡模拟
将得到以下文件:
nvt.log:ASCll文本的日志文件,记录了nvt预平衡过程
nvt.edr:二进制能量文件
nvt.trr:全精度的二进制轨迹文件
nvt.gro:nvt预平衡后的结构
其中在nvt.mdp参数设置索引组



再来看NPT预平衡。
前一步的NVT平衡稳定了体系的温度.在采集数据之前,我们还需要稳定体系的压力(因此还包括密度)。压力平衡是在NPT系综下进行的,其中粒子数,压力和温度都保持不变,这个系综也被称为等温等压系综,最接近实验条件。我们要对体系施加压力直到它达到合适的密度。
正常运行即可。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号