文献绘图--绘制蛋白配体相互作用示意图
原创
文献绘图--绘制蛋白配体相互作用示意图
原创
追风少年i
发布于 2026-05-20 09:00:38
发布于 2026-05-20 09:00:38
作者,Evil Genius
大家学一个组学的内容,一定要全面并且逻辑完整,例如单细胞,从拿到fastq数据开始,自己就能一步一步按照思路往下做,整个形成一套完整的分析逻辑,环环相扣,严丝合缝推出最后的分析结果,千万不要这做一下,那儿做一下,分析的整体观还是没有形成。
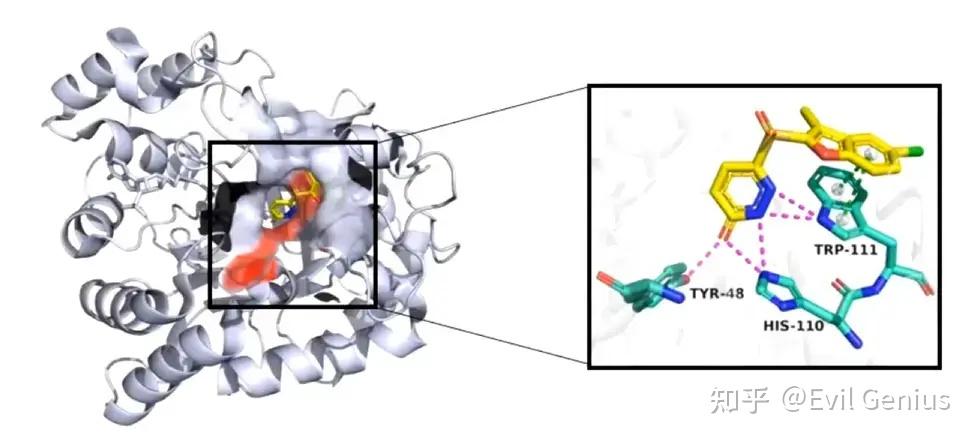
做完所有的分子对接、分子动力学,我们要将结果展示出来放在文献中,类似下面的图
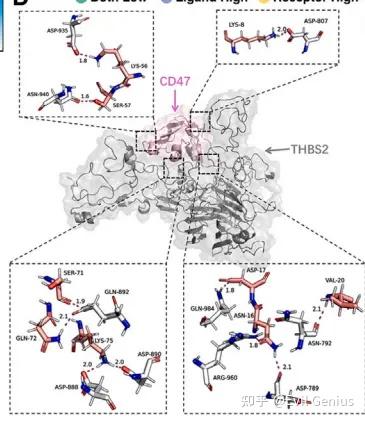
这一篇我们来练习绘图,拿到类似文献中的图
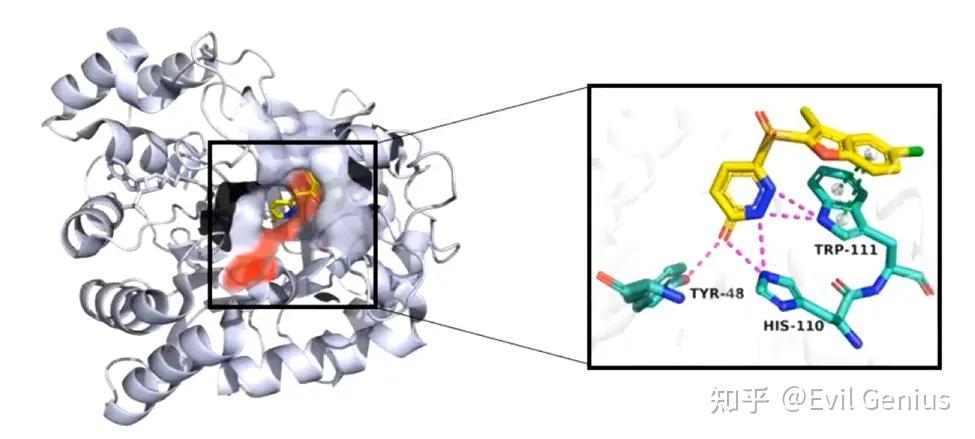
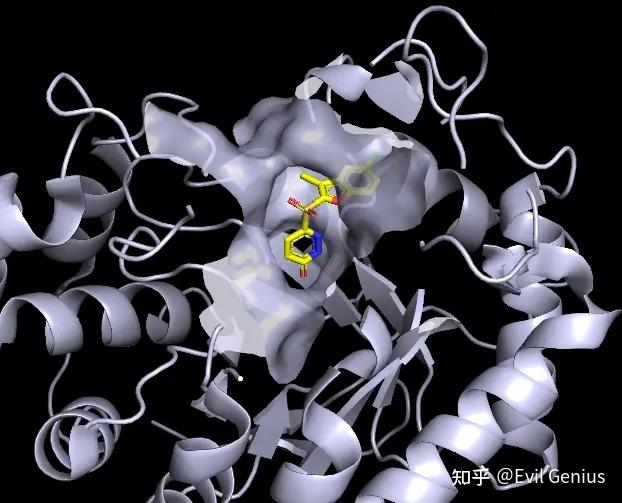
我们先来绘制左边的图。
第一步:去水,去除景深效果,调整蛋白的颜色,之后,调整出序列,调整配体的颜色。
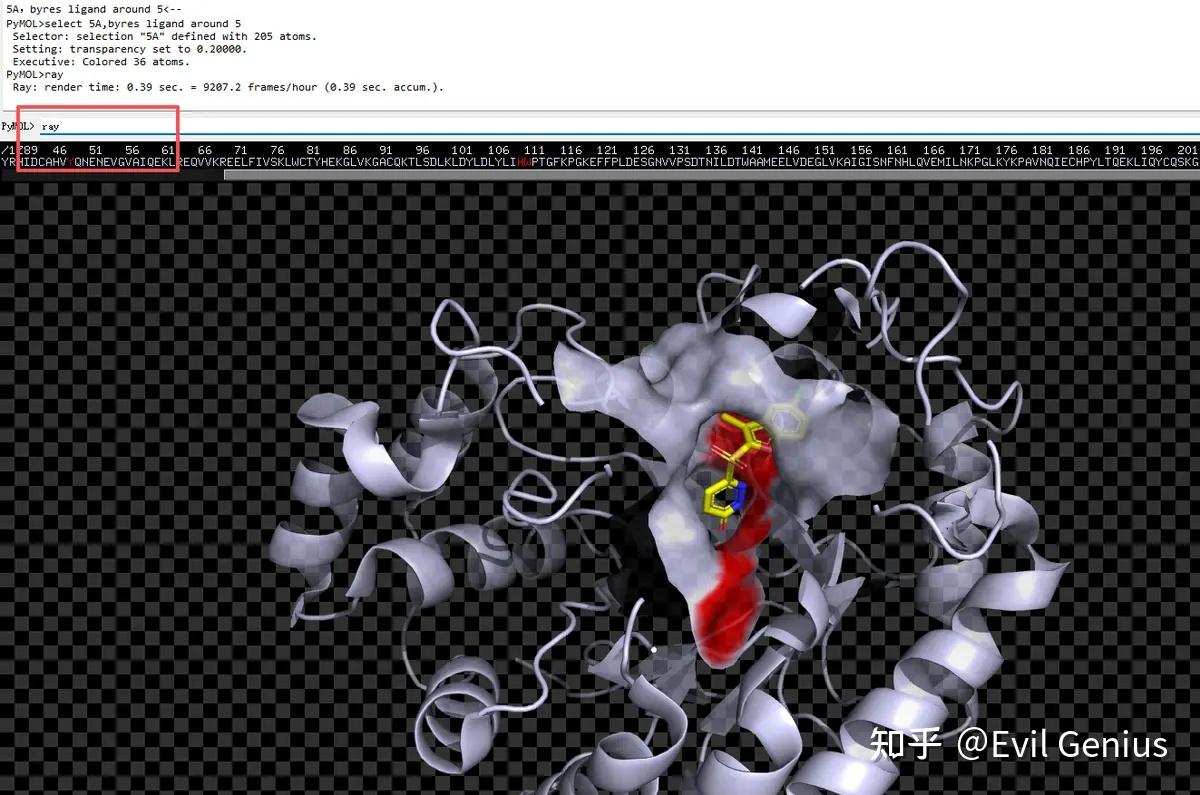
第二步:选择配体,然后将项目命名为“ligand”,选择ligand周围的残基(在命令框中输入“select 5A,byres ligand around 5”)即选中口袋的残基,并且显示为surface
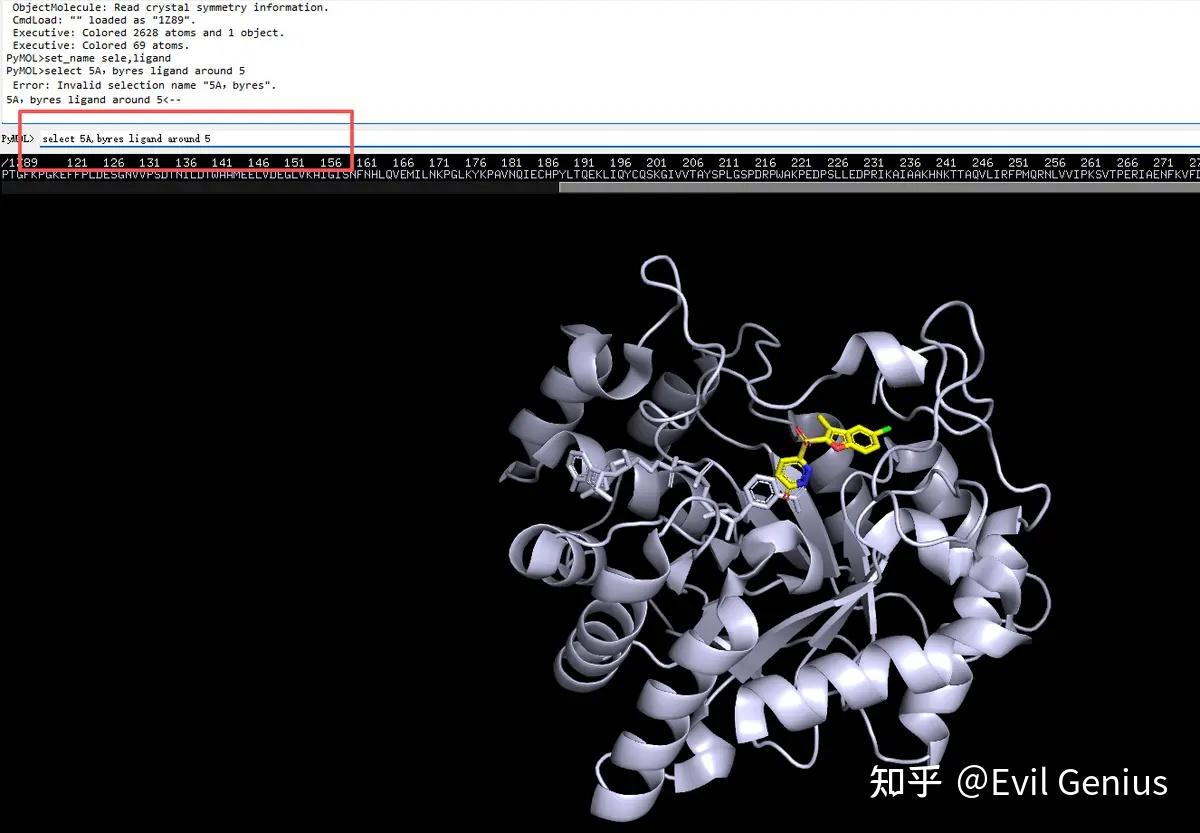
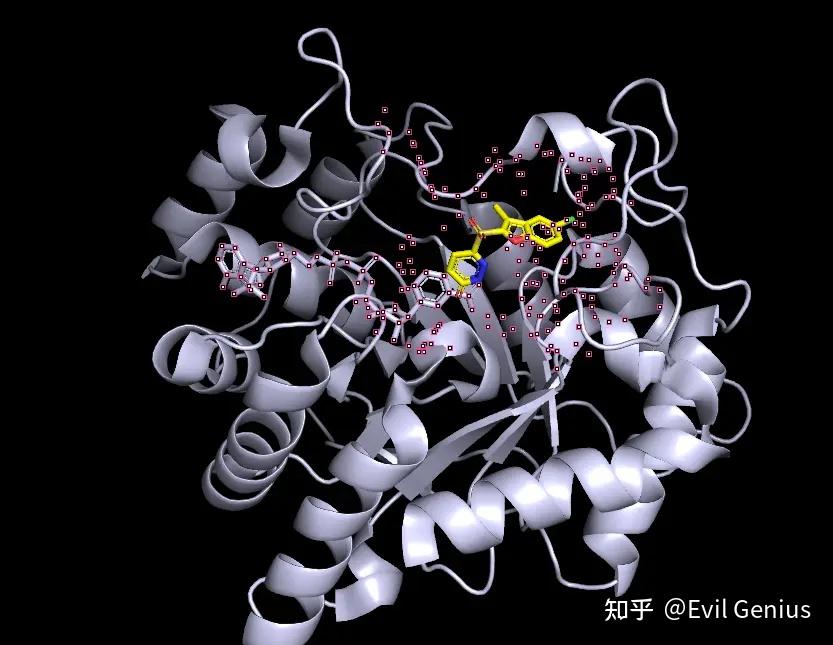
展示为surface
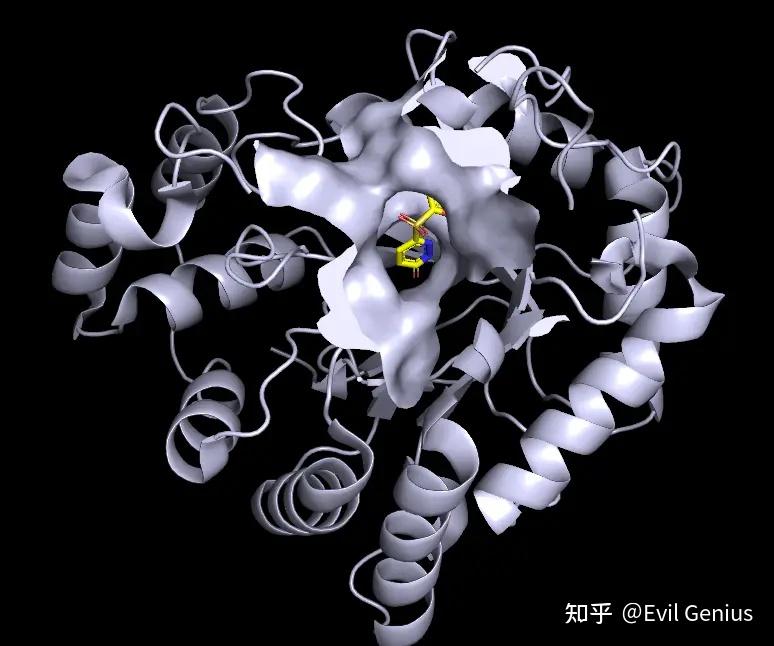
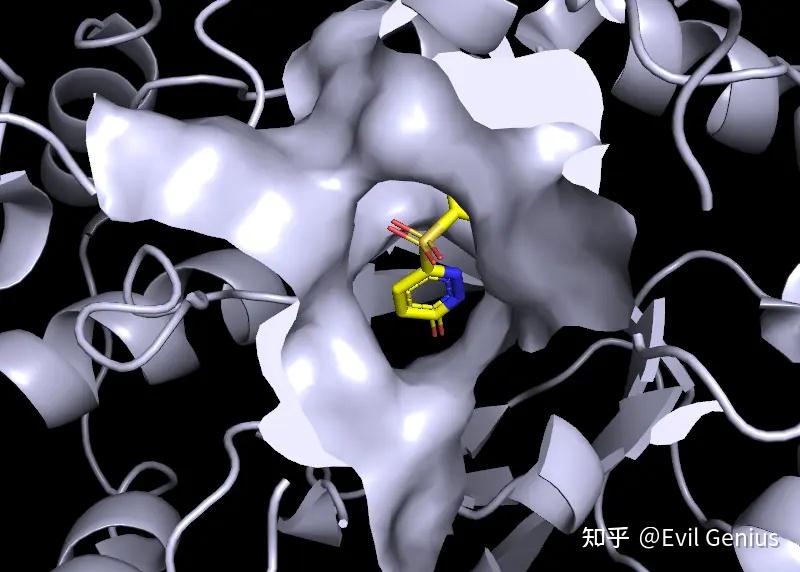
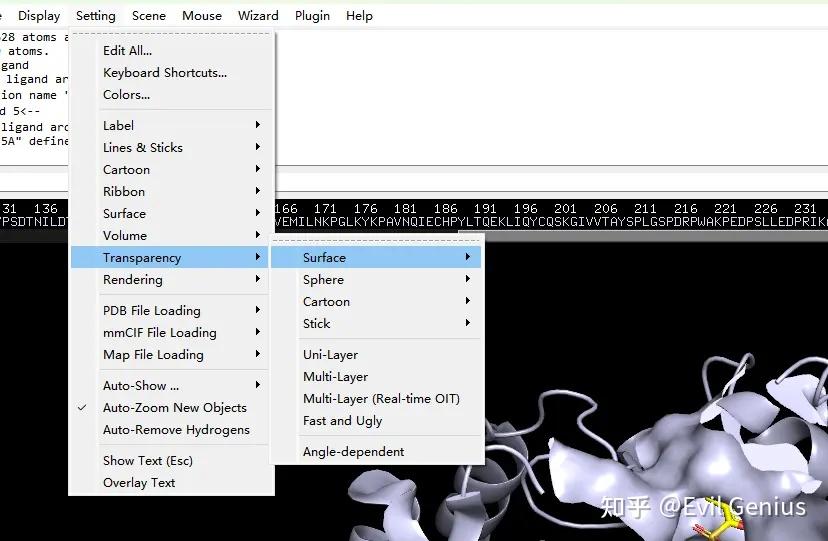
第三步:将surface的透明度设置为20%,将关键的残基(48-110-111)选中并设置为红色
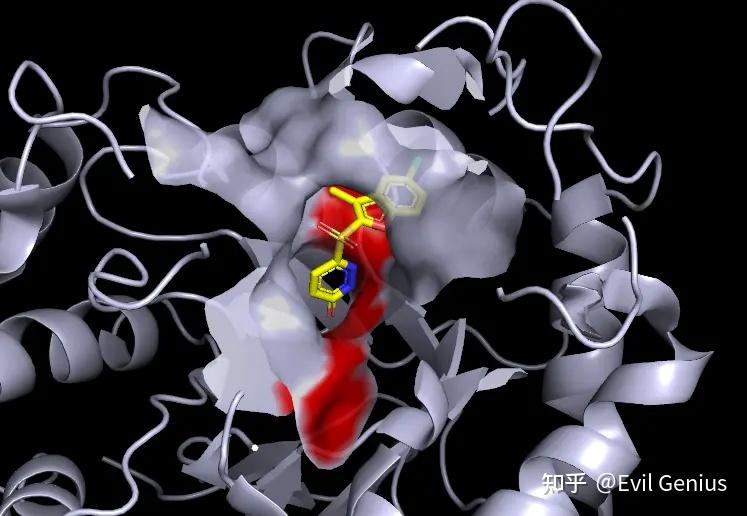
第四步:渲染(ray),保存即可。
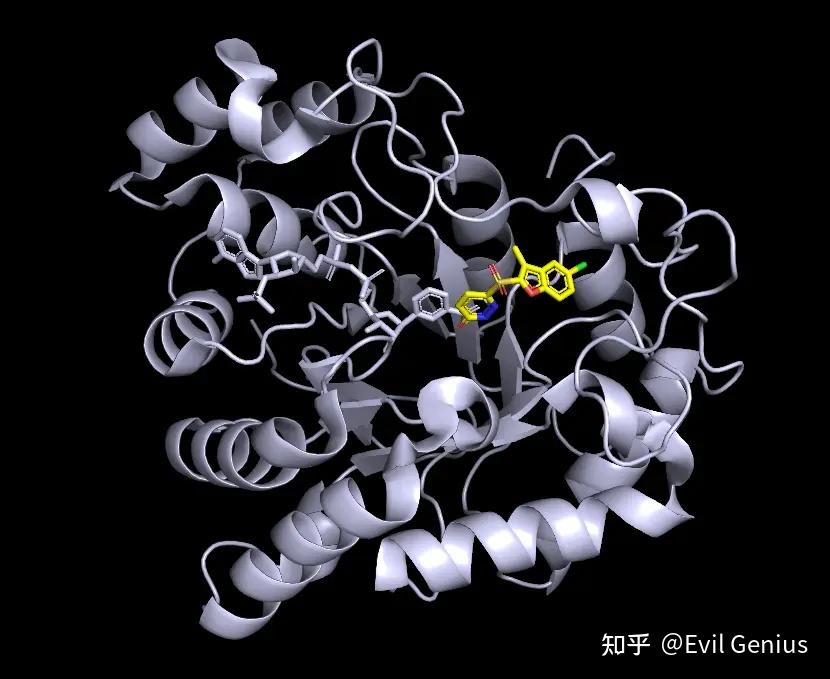
然后第二部分,刚开始一样的操作。
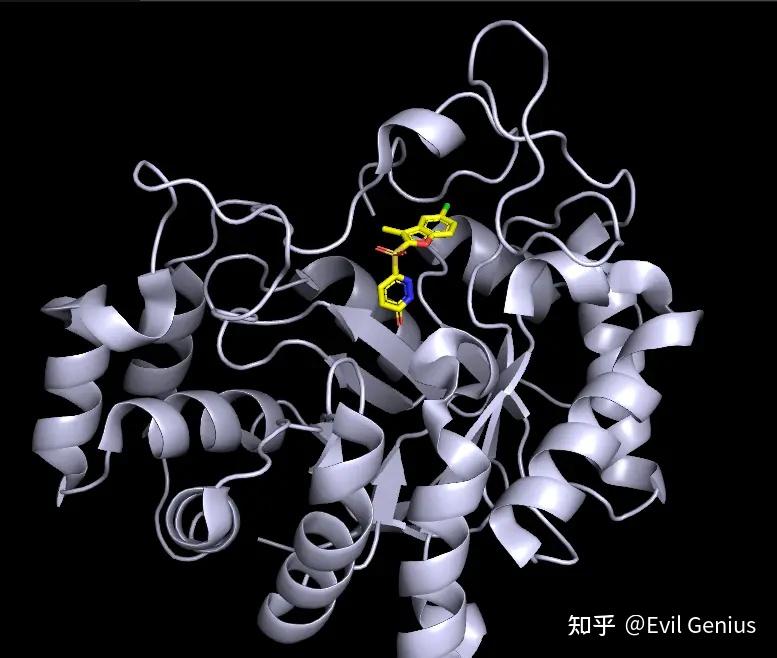
接下来调出配体所在的活性位点
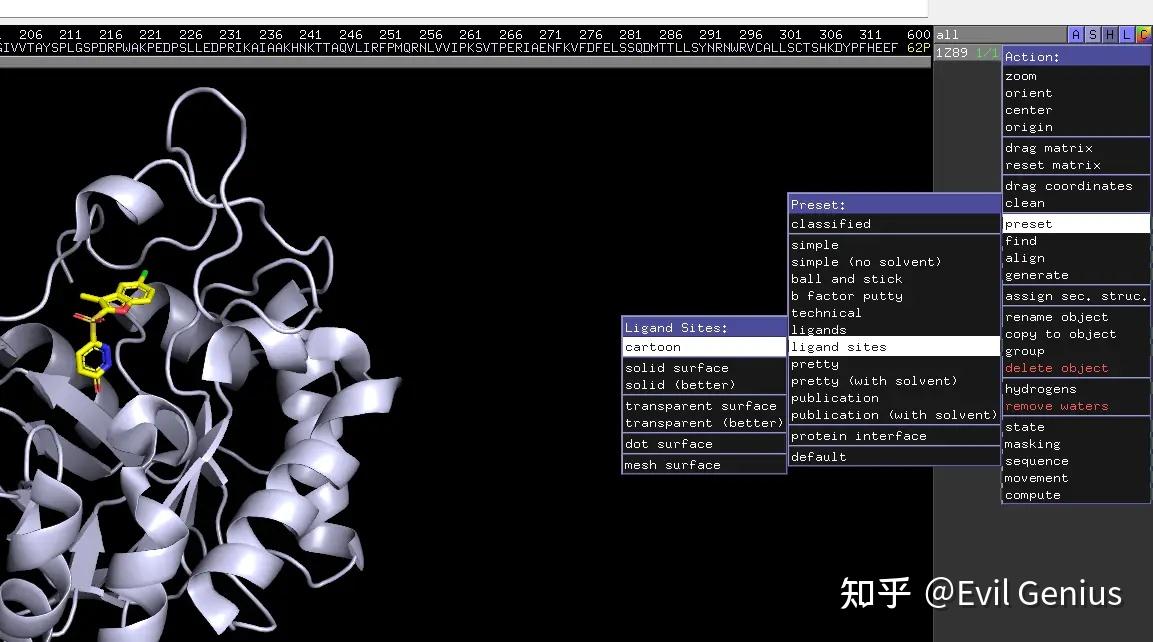
复制项目,新的项目会自动命名为obj01,隐藏1z89的sticks和lines,隐藏obj01的cartoon
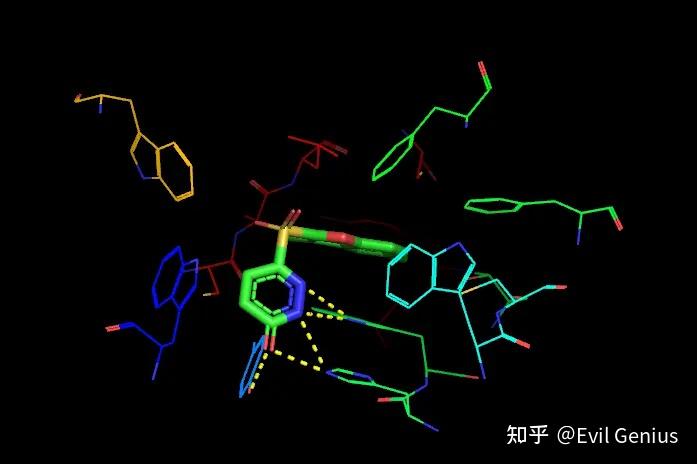
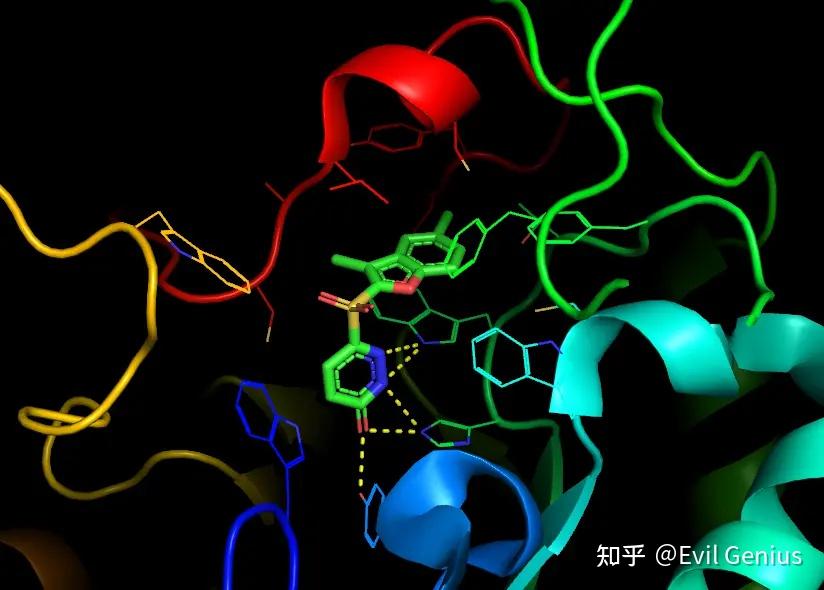
取消显示1z89,界面上鼠标点击选中形成氢键的残基,将他们显示为sticks
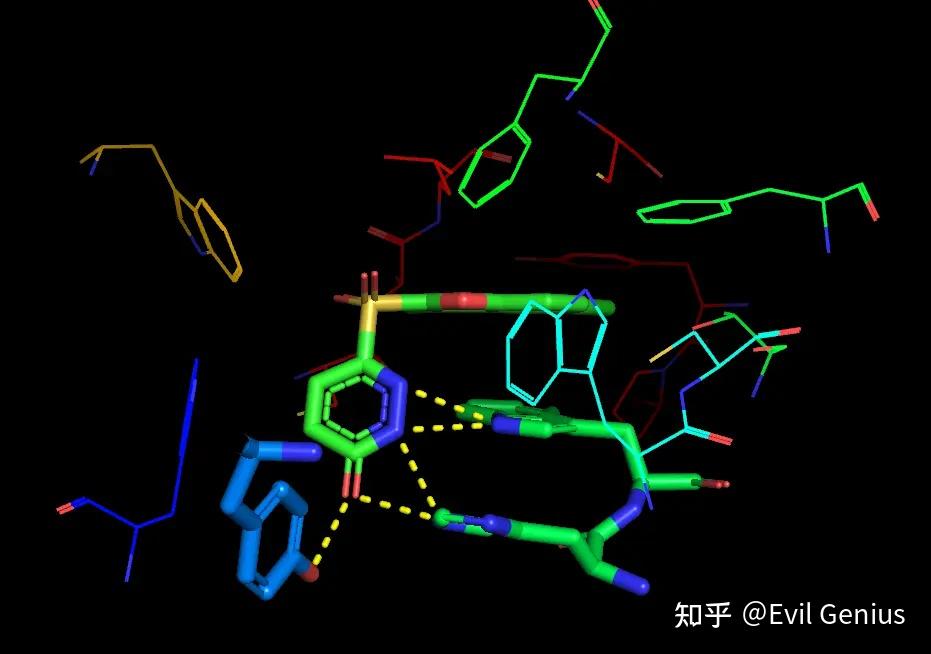
隐藏obj01的lines。将背景改为白色,便于之后的设置
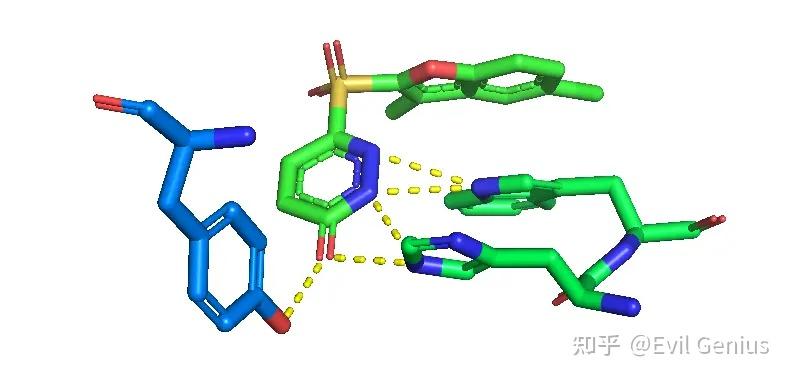
将配体和残基分别设置合适的颜色
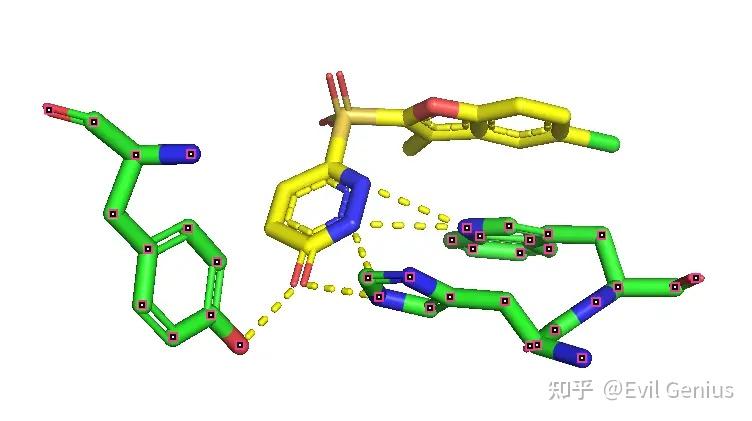
显示π-π相互作用
需要用到的脚本
'''
See more here: http://www.pymolwiki.org/index.php/center_of_mass
DESCRIPTION
Places a pseudoatom at the center of mass
Author: Sean Law
Michigan State University
slaw (at) msu . edu
SEE ALSO
pseudoatom, get_com
'''
from __future__ import print_function
from pymol import cmd
def com(selection, state=None, mass=None, object=None, quiet=1, **kwargs):
quiet = int(quiet)
if (object == None):
try:
object = cmd.get_legal_name(selection)
object = cmd.get_unused_name(object + "_COM", 0)
except AttributeError:
object = 'COM'
cmd.delete(object)
if (state != None):
x, y, z = get_com(selection, mass=mass, quiet=quiet)
if not quiet:
print("%f %f %f" % (x, y, z))
cmd.pseudoatom(object, pos=[x, y, z], **kwargs)
cmd.show("spheres", object)
else:
for i in range(cmd.count_states()):
x, y, z = get_com(selection, mass=mass, state=i + 1, quiet=quiet)
if not quiet:
print("State %d:%f %f %f" % (i + 1, x, y, z))
cmd.pseudoatom(object, pos=[x, y, z], state=i + 1, **kwargs)
cmd.show("spheres", 'last ' + object)
cmd.extend("com", com)
def get_com(selection, state=1, mass=None, quiet=1):
"""
DESCRIPTION
Calculates the center of mass
Author: Sean Law
Michigan State University
slaw (at) msu . edu
"""
quiet = int(quiet)
totmass = 0.0
if mass != None and not quiet:
print("Calculating mass-weighted COM")
state = int(state)
model = cmd.get_model(selection, state)
x, y, z = 0, 0, 0
for a in model.atom:
if (mass != None):
m = a.get_mass()
x += a.coord[0] * m
y += a.coord[1] * m
z += a.coord[2] * m
totmass += m
else:
x += a.coord[0]
y += a.coord[1]
z += a.coord[2]
if (mass != None):
return x / totmass, y / totmass, z / totmass
else:
return x / len(model.atom), y / len(model.atom), z / len(model.atom)
cmd.extend("get_com", get_com)
# vi:expandtab:sw=3先运行这个脚本
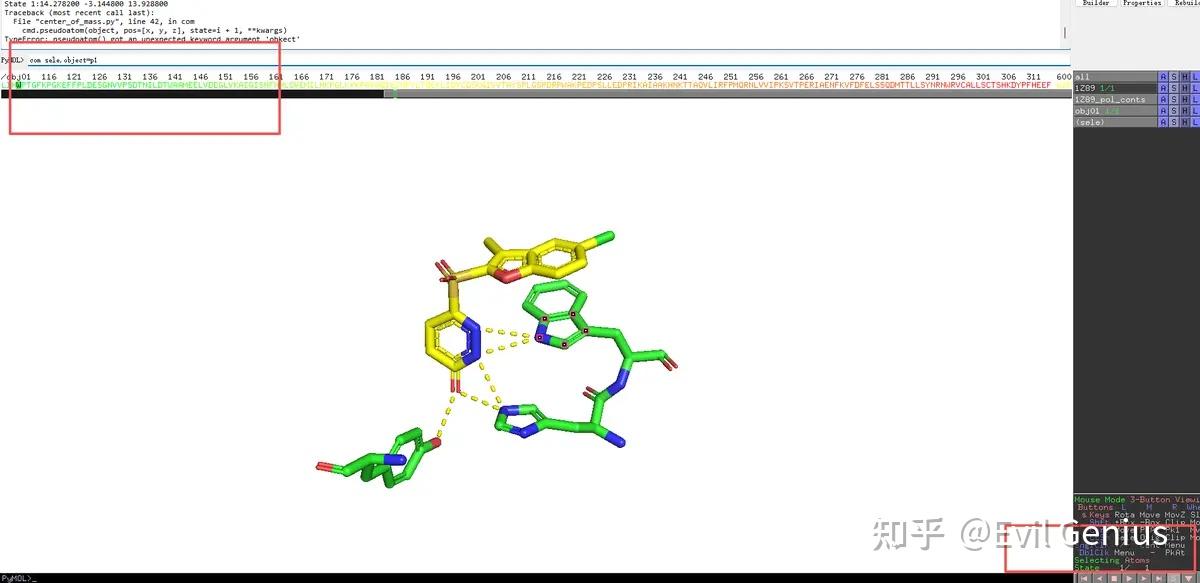
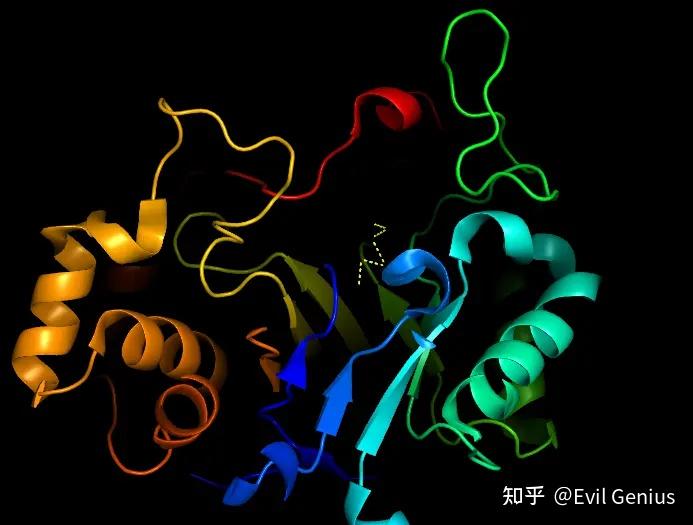
多了一个球
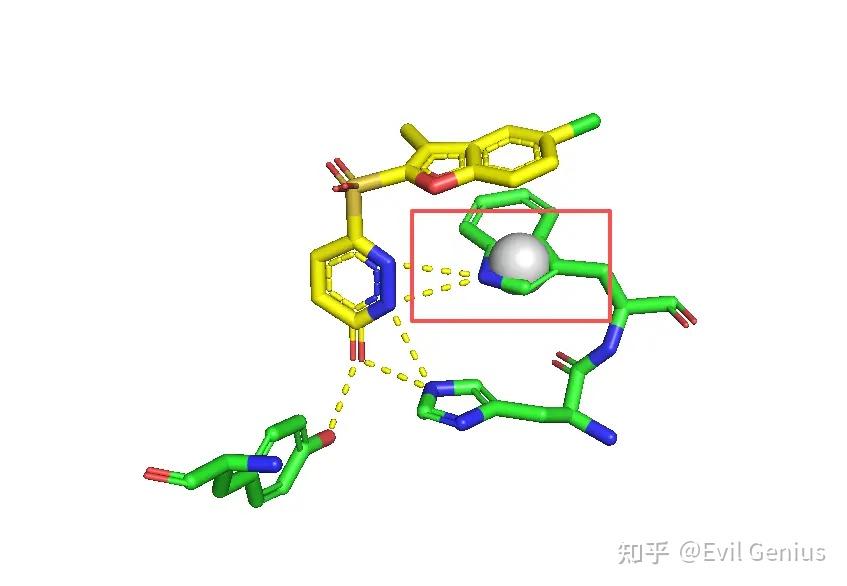
同样的方式
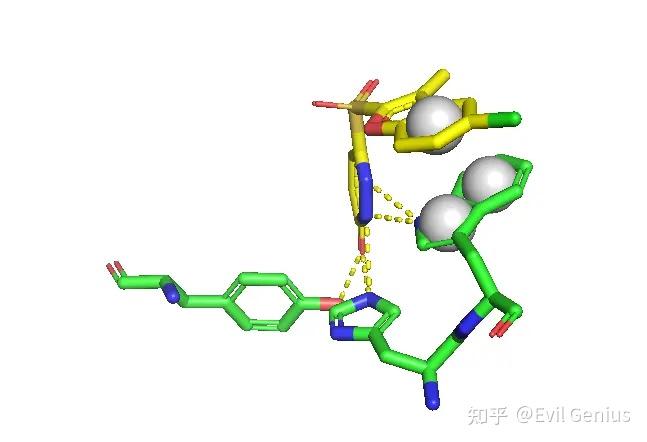
调整球的大小
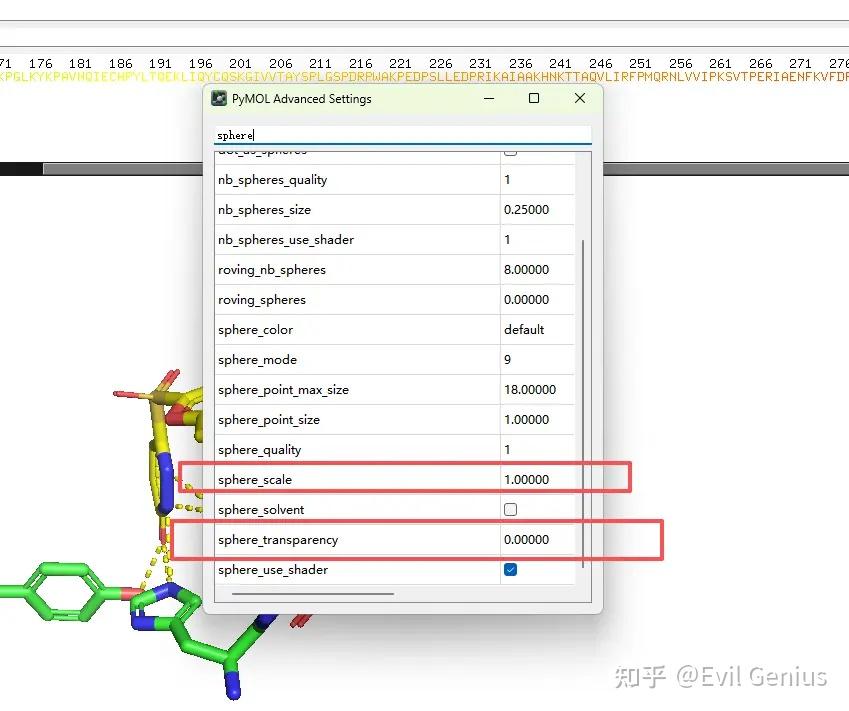
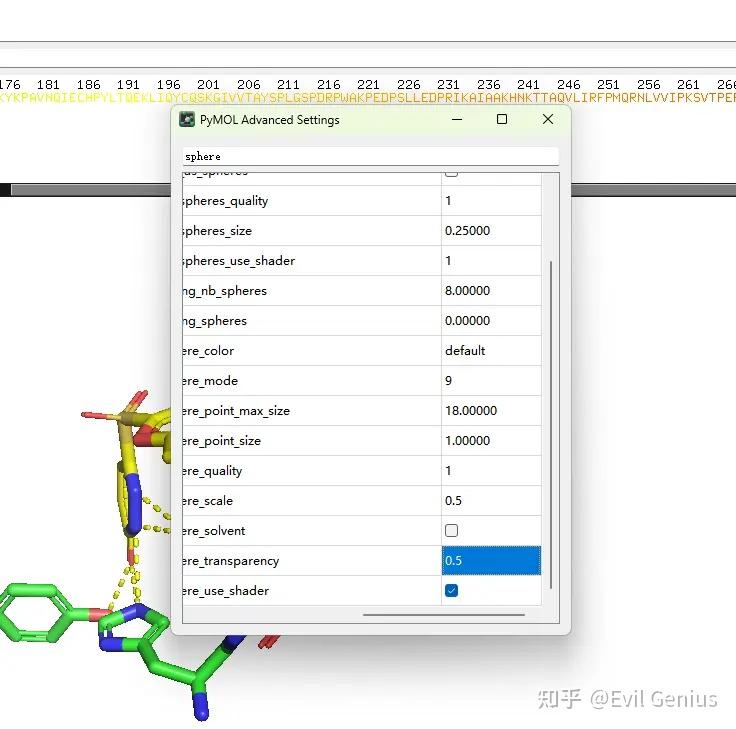
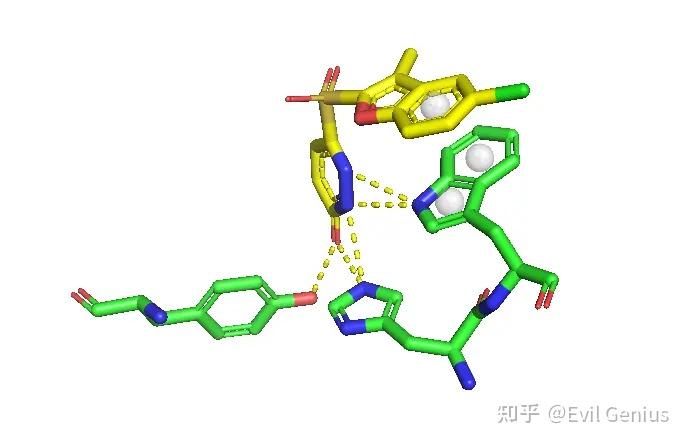
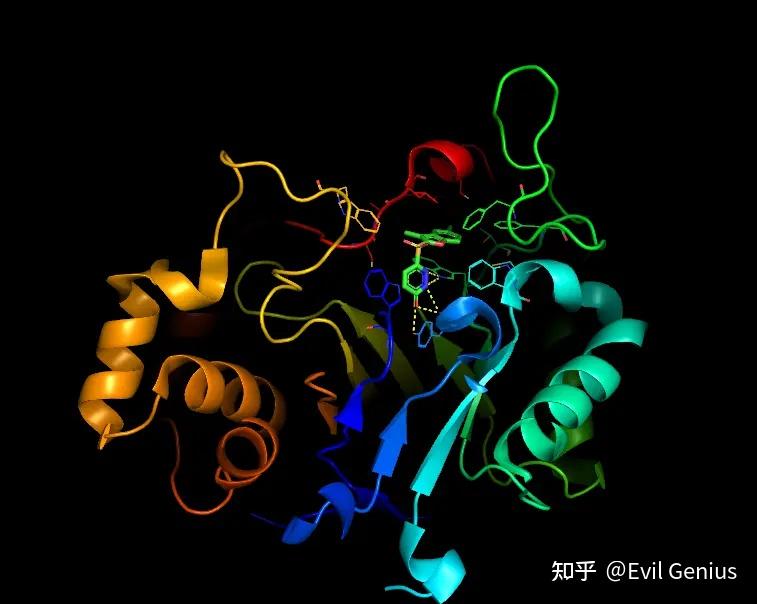
添加π-π相互作用
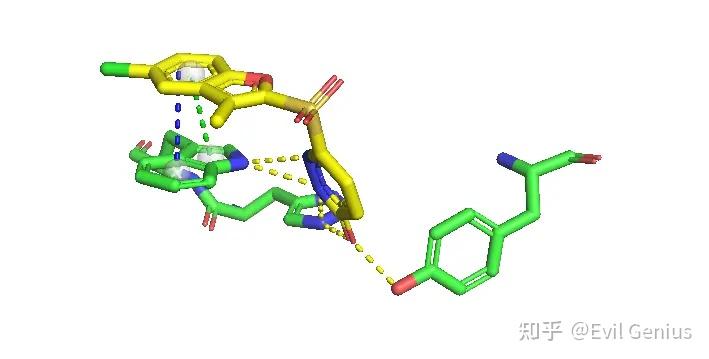
添加标签
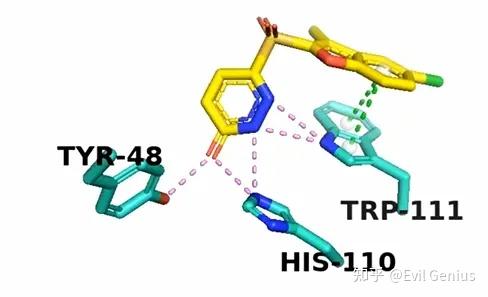
最后精细化调整一下
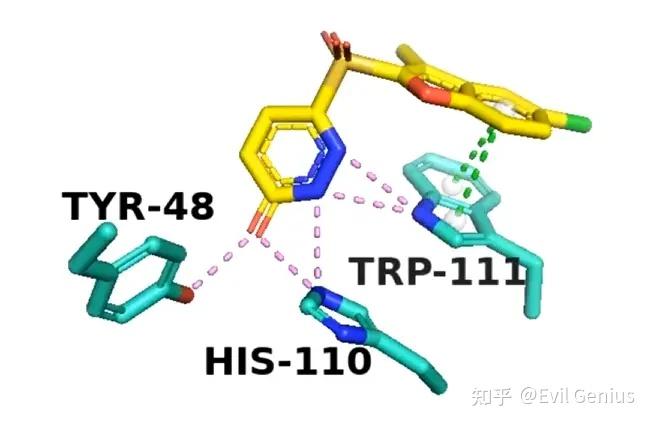
渲染,保存,组图
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号