AI驱动蛋白设计中经过湿实验验证的方法全景

AI驱动蛋白设计中经过湿实验验证的方法全景

DrugIntel
发布于 2026-05-26 19:53:12
发布于 2026-05-26 19:53:12

文献来源:Current Opinion in Structural Biology 2026, 98: 103272 DOI:10.1016/j.sbi.2026.103272 作者:Clayton W. Kosonocky¹, Sarah Alamdari², Kevin K. Yang², Ava P. Amini² 机构:¹ 德克萨斯大学奥斯汀分校;² Microsoft Research(剑桥)
导读:为什么这篇综述值得精读?
近年来,AI蛋白质设计领域论文产出呈爆炸式增长,但绝大多数工作止步于计算层面的评估——模型生成的序列是否真的在实验室中起效,往往语焉不详。这篇综述恰好填补了这一空白:它系统梳理了横跨完整"数据→模型→实验"流程、并经过实验验证的AI蛋白质设计方法,提供了迄今为止最为全面的实验成功率参考数据库。
一、背景:从传统方法到AI驱动的范式转变
蛋白质设计的核心挑战在于导航氨基酸序列、三维结构与生物功能之间的复杂映射关系。
1.1 传统方法的局限
传统计算蛋白质设计依赖:
- • 物理模拟(分子动力学、量子力学/分子力学)
- • 经验打分函数(如 Rosetta 能量函数)
- • 专家启发式规则
这些方法的主要瓶颈包括:计算成本高昂、难以大规模扩展、高度依赖高质量结构信息,以及对序列空间的采样效率低。
1.2 AI方法的优势
AI模型通过从大规模生物数据中直接学习序列—结构—功能关系,绕过了上述限制:
数据规模 | 资源 | 数量级 |
|---|---|---|
蛋白质结构 | 蛋白质数据库(PDB) | 数十万条 |
蛋白质序列 | UniRef / UniProt | 数亿至数十亿条 |
正是这种数据规模的优势,使得深度学习模型能够捕捉传统方法难以建模的统计规律。
二、端到端设计流程详解
作者将AI驱动蛋白质设计拆解为四个相互依存的核心环节:
数据集构建 ──▶ 模型训练 ──▶ 候选生成与筛选 ──▶ 实验验证
▲ │
└──────── 实验反馈回路 ◀──────────┘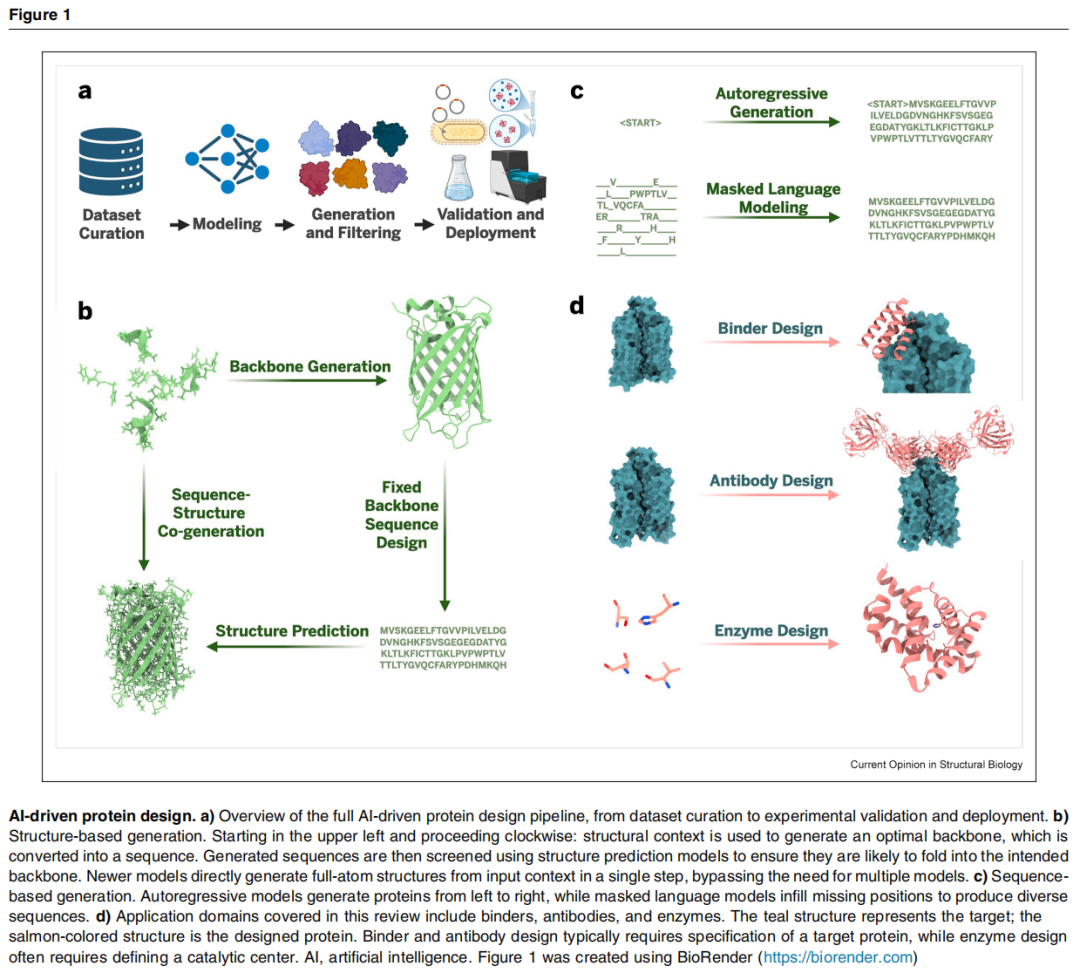
2.1 数据集构建
数据质量是整个流程的根基。关键考量包括:
- • 序列多样性:覆盖蛋白质序列空间的广度,影响模型的泛化能力
- • 结构注释质量:高分辨率结构(X射线晶体学、冷冻电镜)vs. 同源建模结构的权衡
- • 功能标注:结合数据(Kd值)、酶活数据等功能信息的覆盖程度
- • 数据偏差:PDB中小分子配体代表性不足,是蛋白质—小分子结合蛋白设计成功率偏低的根本原因之一
2.2 模型架构:两大技术路径
路径一:基于结构的方法(Structure-Based)
结构背景输入
│
▼
骨架生成模型(Backbone Design)
[RFdiffusion, Chai-2, LatentX...]
│
▼
固定骨架序列设计(Fixed-Backbone Sequence Design)
[ProteinMPNN, LigandMPNN, CARBonAra...]
│
▼
结构预测验证与筛选
[AlphaFold2, Boltz-1, ESMFold...]条件输入的演进:
- • 早期模型:仅接受残基坐标或骨架原子
- • 近期模型:支持全原子生物分子表示(包括小分子、核酸)及动态构象状态
路径二:基于序列的方法(Sequence-Based)
序列语言模型完全绕过结构信息,直接在序列空间中操作:
- • 自回归模型(如 ProGen2, ProGen3):从左到右逐步生成序列
- • 掩码语言模型(如 EvoDiff, PepMLM):通过填充缺失位置生成多样序列
- • 条件生成策略:在部分序列、对齐同源序列、或特定酶家族分布上条件化生成
值得关注的发现:序列语言模型在结合蛋白设计(传统上被视为结构任务)上展现出竞争力,可能源于模型从协同进化信号中隐式学习到了结构约束。
统一方法与混合策略
部分模型打破了结构/序列的二元划分:
- • 序列—结构联合生成(Chroma, RoseTTAFold Sequence Space Diffusion):同步生成骨架与序列
- • 可微分结构预测器反向传播(BindCraft, Germinal):通过梯度优化满足生物/物理约束
- • 全原子设计(RFdiffusion All-Atom, RFdiffusion3):直接在全原子层面建模生物分子相互作用
2.3 候选生成与筛选
典型的筛选流程(按计算成本递增):
- 1. 结构预测置信度过滤:丢弃 pLDDT / pTM 低于阈值的候选
- 2. Rosetta 能量打分:剔除能量不利或非物理结构
- 3. 序列/结构多样性聚类:维持候选集的多样性,避免冗余实验
- 4. 人工检查:对关键位点(结合界面、催化残基)的人工审核
2.4 实验验证
验证的层次结构(分辨率递增,通量递减):
验证层次 | 方法 | 目的 |
|---|---|---|
表达验证 | SDS-PAGE, SEC | 确认正确尺寸、单分散性、无聚集 |
二级结构 | 圆二色谱(CD) | 快速评估折叠状态 |
结合亲和力 | SPR, BLI, YSD | 定量测定 Kd、kon、koff |
原子级结构 | X射线晶体学、冷冻电镜 | 验证结合模式的金标准 |
三、三大应用领域深度解析
3.1 结合蛋白(Protein Binders)
设计挑战
结合蛋白设计要求模型精确捕捉蛋白质—靶标界面的互补性,需要同时优化形状互补、静电相互作用和疏水效应。
主流方法与代表模型
方法策略 | 代表模型 | 核心思路 |
|---|---|---|
扩散骨架生成 | RFdiffusion, Chai-2, LatentX | 以靶标表面为条件,扩散生成兼容骨架 |
可微分优化 | BindCraft, Germinal | 反向传播梯度优化序列满足结构约束 |
表面指纹匹配 | dMaSIF | 学习几何表面特征,匹配互补界面 |
序列语言模型 | PepMLM, EvoDiff | 在靶标条件下生成结合肽/蛋白序列 |
多聚体结构预测 | AlphaProteo, PXDesign | 利用多聚体结构预测模型框架设计 |
关键实验数据
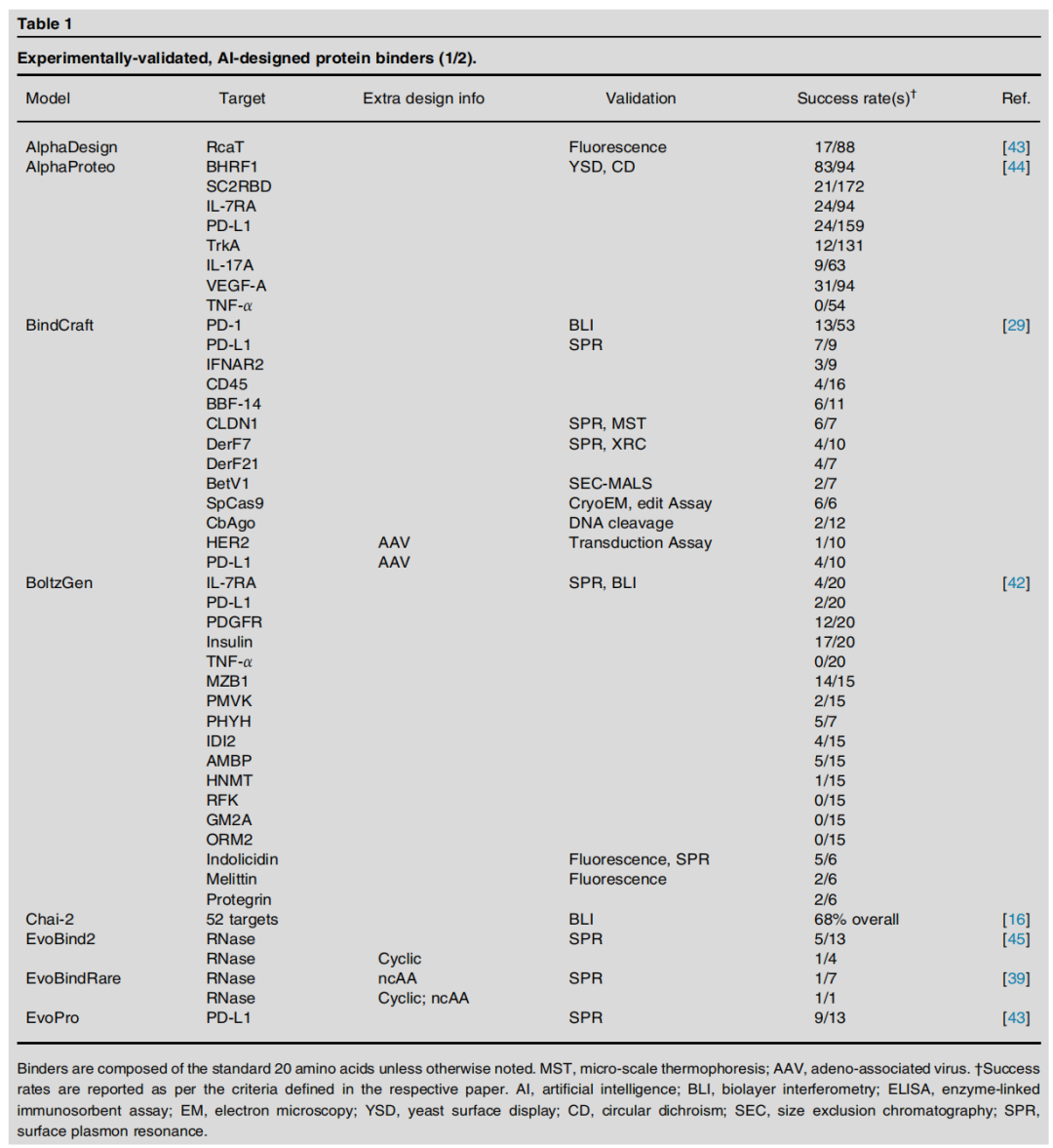
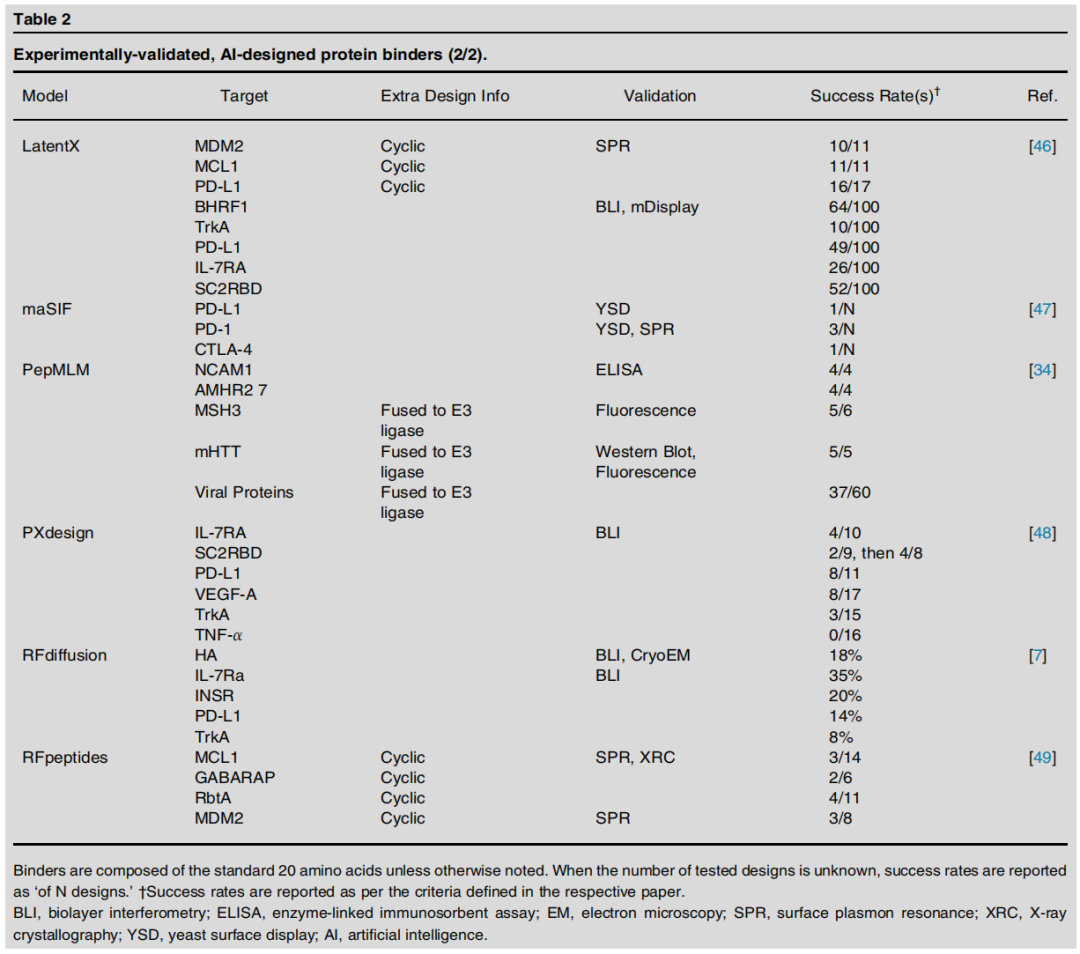
TNF-α 的特殊性:在所有测试模型中,仅 Chai-2 成功生成了 TNF-α 结合蛋白,揭示了该靶标界面的独特挑战性(如高度平坦的结合表面)。
蛋白质—小分子结合蛋白的困境
尽管 RoseTTAFold All-Atom、LigandMPNN、LASErMPNN 等模型已展示了蛋白质—小分子设计的初步成功,但成功率普遍低于蛋白质—蛋白质结合蛋白。根本原因在于:PDB中小分子配体的化学多样性覆盖极为有限,相对于真实化学空间的广度,训练数据严重不足。
当前局限
- • 大多数验证基于体外结合,缺乏细胞或动物模型中的功能验证
- • 免疫原性控制是治疗应用的核心挑战(非自身表位、异常结构基序可引发免疫反应)
- • 脱靶结合(特别是在复杂生物环境中)亟需更系统的评估
3.2 抗体设计(Antibody Design)
独特挑战
抗体设计面临的技术障碍与一般结合蛋白存在本质差异:
数据层面:
- • 尽管 SAbDab 收录了几乎全部已知抗体结构,PDB中抗体结构覆盖度依然有限
- • Observed Antibody Space、AbRank 等序列库收录数亿条序列,但仅小部分有原子分辨率结构
- • V(D)J 重组 + 体细胞超突变产生的 CDR H3 极端序列多样性,导致个体抗体几乎无深度同源家族
模型层面:
- • 基于多序列比对(MSA)的方法(AlphaFold2风格)在抗体上表现受限——协同进化信号稀疏
- • CDR环的构象多样性超出通用蛋白质折叠模型的建模能力
- • 结构基础扩散模型倾向于生成α螺旋主导的CDR环(与真实无序CDR环不符)
技术突破:引入抗体专属生物先验
近年关键进展在于将领域专属知识注入模型设计:
- • Germinal:引入自定义损失函数,惩罚模型生成α螺旋CDR结构,引导生成更真实的无序CDR环;同时支持表位定向设计
- • IgGM:支持框架区工程化、亲和力成熟(结合界面系统优化)与人源化(减少免疫原性)的一体化设计流程
- • mBER:百万级实验筛选支持的可控从头抗体设计,实现对145个靶标的广覆盖
- • RFantibody:原子级精确度的抗体设计,在病毒靶标上通过冷冻电镜验证
关键实验数据
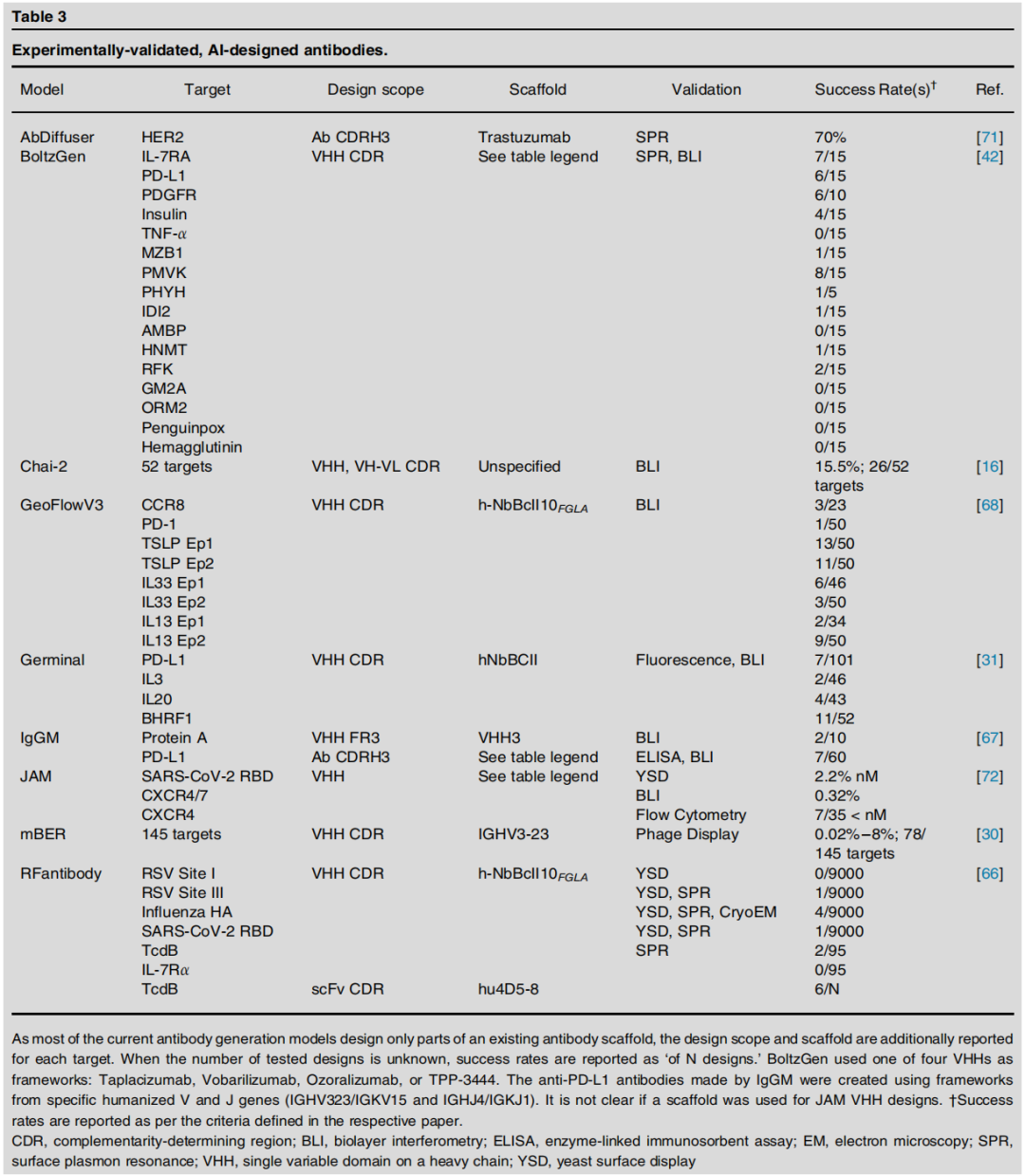
治疗开发视角
AI抗体设计正从 CDR 重设计向全流程设计演进,但以下属性仍面临数据限制:
- • 非蛋白质抗原(聚糖、脂质、核酸)的识别设计
- • 高精度人源化(人源化程度 vs. 亲和力的权衡)
- • 可开发性(aggregation propensity, thermal stability, viscosity)
3.3 酶设计(Enzyme Design)
为何最难?
酶设计在三个应用方向中挑战最大,因为它要求模型同时建模:
- 1. 蛋白质本身的折叠与稳定性
- 2. 底物(往往是 PDB 中代表性不足的小分子)
- 3. 催化机制:过渡态几何构型、活性位点静电环境、催化残基精确定向
- 4. 动力学:需要多种构象状态,而非单一静态结构
催化涉及瞬态、高能量中间体,这些信息难以从静态结构中直接推断,可能需要结合量子力学/分子力学(QM/MM)、密度泛函理论(DFT)等物理方法提供先验。
方法演进路线
早期方法
├── 幻觉法(Hallucination):trRosetta 反向传播设计荧光素酶
└── 骨架扩散 + 固定催化残基:PLACER
│(限制:催化位点构象固定,设计空间受限)
▼
近期方法
├── 学习最优催化残基定位:RFdiffusion2
├── 全原子生物分子相互作用设计:RFdiffusion3
├── 催化基序骨架化:RiffDiff
└── 序列语言模型条件化生成:ZymCTRL, ProGen2, ProtGPT2关键实验数据
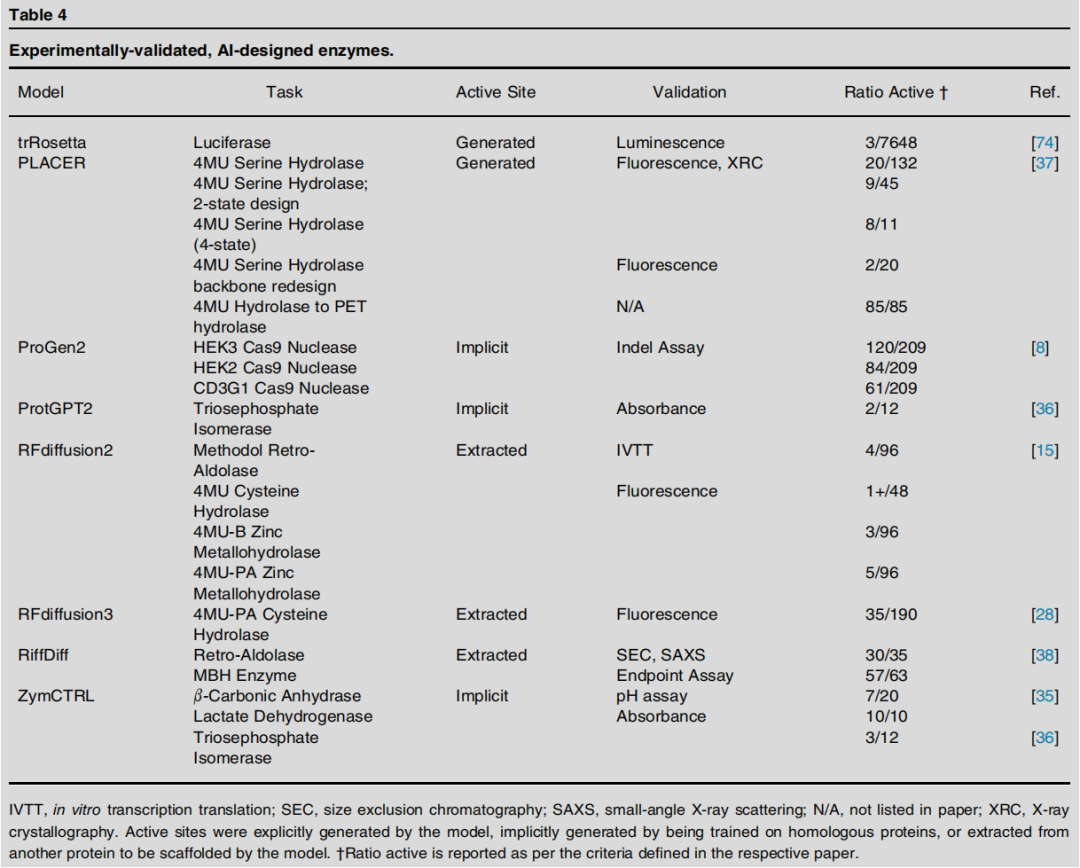
展望:迈向任意催化机制
现有AI酶设计工具所覆盖的催化反应种类,仍仅是自然界已知酶反应的极小子集。突破方向在于:
- • 将 DFT 计算的过渡态几何作为设计约束(RFdiffusion2 已开始支持)
- • 整合分子动力学模拟(BioEmu、Boltzmann Generators)建模构象动态
- • 开发可将催化活性转化为可测量信号的通用高通量实验平台
四、跨应用领域的横向比较
维度 | 结合蛋白 | 抗体 | 酶 |
|---|---|---|---|
数据基础 | PDB复合物结构(丰富) | SAbDab结构 + 序列库 | PDB + 底物信息(稀缺) |
最高实验成功率 | ~88%(BHRF1) | ~70%(HER2 CDRH3) | ~100%(特定任务) |
结构验证 | 多个复合物 CryoEM/XRC | 少量CryoEM验证 | XRC(少数) |
通量瓶颈 | SPR/BLI可支持高通量 | 与结合蛋白相近 | 底物特异性实验,难以标准化 |
临床转化壁垒 | 免疫原性、体内特异性 | 人源化、可开发性 | 底物范围、稳定性 |
AI方法成熟度 | 最高 | 高,快速发展中 | 相对早期 |
五、核心论断与方法论启示
5.1 实验反馈是核心驱动力
文章最重要的论断之一:
进展的根本驱动力不是模型规模,而是实验数据的质量与覆盖度。
结合蛋白设计之所以进展最快,并非因为模型最复杂,而是因为 SPR/BLI 等结合实验高度可扩展,能够为模型提供大规模定量反馈。相比之下,酶设计因底物多样、缺乏标准化实验平台,进展相对分散。
5.2 数据稀缺性的具体体现
- • 蛋白质—小分子结合:小分子化学空间巨大,PDB覆盖率极低
- • 脱靶特异性:缺乏系统性的脱靶结合测量数据
- • 人源化:精确人源化需要大量配对的免疫原性测量数据
- • 酶底物多样性:不同底物需要完全不同的实验验证流程
5.3 "闭环"的真正含义
文章标题"Closing the Loop"不只是比喻:
计算假设 ──▶ 实验验证 ──▶ 数据反馈 ──▶ 模型迭代改进
▲ │
└────────────────────────┘当前的主要断点:
- 1. 大多数实验验证为体外结合,缺乏生理相关环境中的功能验证
- 2. 成功数据共享不成问题,但失败数据(阴性结果)的系统收集与共享严重不足
- 3. 设计规模(数十到数百候选)与实验通量的匹配仍是瓶颈
六、未来方向与开放挑战
近期可期的突破点
- • 全原子建模的普及:RFdiffusion3、AtomWorks 等全原子模型将进一步降低蛋白质—小分子和蛋白质—核酸设计的门槛
- • 高通量实验平台:开发将更多功能属性(酶活、选择性、稳定性)转化为可扩展测量信号的实验方案
- • 抗体人源化与可开发性:将人源化和可开发性评估整合进设计循环
中长期挑战
- • 酶催化机制扩展:将 QM/MM 与 DFT 计算的先验知识系统集成到扩散/语言模型
- • 体内功能验证:从体外结合到细胞水平、动物模型的验证能力建设
- • 设计的可解释性:理解模型为何生成特定设计,而非仅追求黑盒优化
- • 多靶标与多功能设计:如广谱抗病毒、泛癌症靶向
小结
这篇综述的价值在于用实验数据说话。它提醒我们:在AI蛋白质设计领域,模型架构的创新固然重要,但实验反馈的质量、数量与多样性才是决定进步速度的真正瓶颈。
未来的突破不会来自单一的计算突破,而将来自计算与实验的深度协同——一个持续运转、不断校准的"闭合回路"。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-24,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号