分子动力学--分析结果的详细解读
原创
分子动力学--分析结果的详细解读
原创
追风少年i
发布于 2026-05-29 10:08:26
发布于 2026-05-29 10:08:26
作者,Evil Genius
这一篇我们来详细解读分子动力学最后的分析结果。
首先第一个:RMSD--用于量化分子结构的变化程度或差异大小。
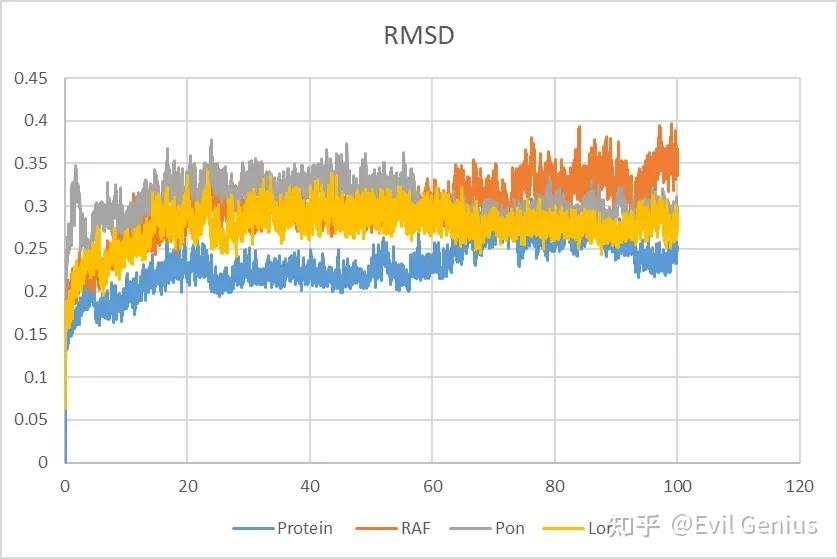
RMSD的定义:均方根偏差计算的是两个分子构象(例如,模拟中某一时刻的结构与参考结构)之间对应原子位置差异的平均值。
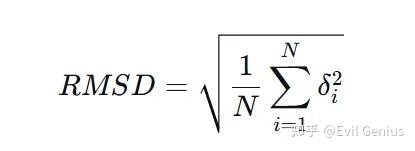
N:用于比较的原子数量(一般我们比较的是C骨架)
δ i :第 i个原子在两个结构中的坐标差
主要用途
用途 | 说明 |
|---|---|
评估平衡 | RMSD随时间趋于稳定,说明体系已达到平衡 |
比较构象 | 折叠/结合后结构是否接近天然态 |
验证稳定性 | 药物与靶点结合后RMSD低,说明复合物稳定 |
聚类分析 | 根据两两间RMSD区分不同构象状态 |
参考结构:通常选X射线晶体结构、模拟起始结构或平均结构
原子选择:
全原子(C、N、O、S等)—— 对构象变化敏感
主链/骨架(常用)—— 评估蛋白整体折叠状态
Cα原子 —— 适用于蛋白质
重原子 —— 适用于小分子/配体
对齐(拟合):计算前必须先通过刚体旋转/平移(如Kabsch算法)使两个结构尽可能重叠,否则RMSD会被整体分子朝向差异主导
结果解读
蛋白质
RMSD(主链) | 含义 |
|---|---|
< 1 Å | 几乎相同(结晶状态差异) |
1–2 Å | 很相似(热波动或微小运动) |
2–3 Å | 明显构象变化 |
> 3 Å | 很大差异(可能完全不同构象或未折叠) |
小分子配体
RMSD | 含义 |
|---|---|
< 0.5 Å | 非常稳定,结合模式一致 |
0.5–1.5 Å | 一定柔性,仍处在结合口袋内 |
> 2 Å | 可能漂移出结合位点或翻转 |
常见分析图
时间序列图(RMSD vs. 时间):
初始阶段快速上升 → 体系弛豫
趋于平台波动 → 达到平衡
持续大幅上升 → 失稳/变性/崩溃
直方图/分布图:
单峰 → 主要在单一构象状态
多峰 → 多个亚稳态间跳跃
2D RMSD图(所有构象两两计算):
能清晰看出“状态-状态”转化路径和簇。
第二个:Rg(回转半径)是分子动力学模拟中衡量分子结构紧凑程度或空间扩展程度的重要几何参数。
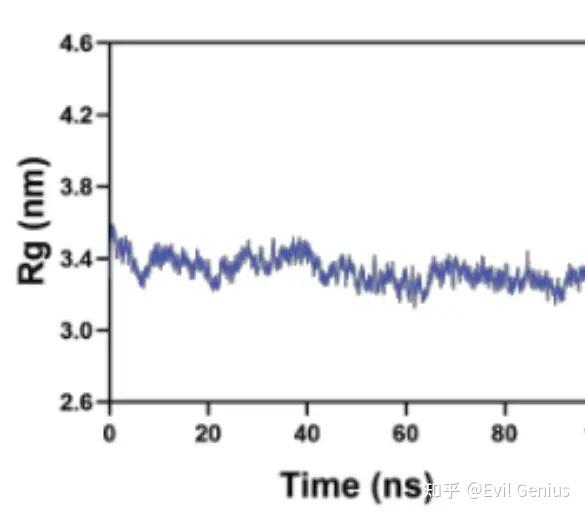
Rg:回转半径定义为分子中所有原子到其质心的距离的均方根值:
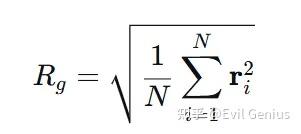
其中 ri是原子 i 到分子质心的向量。
Rg 值小 → 原子密集地包裹在质心周围 → 结构紧凑(折叠态、球状)
Rg 值大 → 原子远离质心扩散 → 结构松散(伸展态、变性态)
主要分析目标
用途 | 说明 |
|---|---|
监测蛋白质折叠/去折叠 | 折叠时 Rg 减小,去折叠时 Rg 增大 |
评估结构稳定性 | 平衡阶段 Rg 在均值附近稳定波动 → 结构稳定 |
检测构象转变 | Rg 突然跳跃 → 发生了折叠/膨胀/坍塌事件 |
区分不同构象状态 | 结合 RMSD,可判断构象变化是紧凑性变化还是重排 |
验证实验数据 | 与 SAXS(小角X射线散射)测得的实验 Rg 对比验证模拟可靠性 |
典型数值范围
蛋白质(取决于大小和形状):
状态 | Rg 相对变化 | 示例(约 100 残基蛋白) |
|---|---|---|
完全折叠(天然态) | 小(紧凑) | ~15–18 Å |
部分去折叠 | 中等 | ~20–30 Å |
完全去折叠(无规卷曲) | 大(伸展) | ~40–60 Å |
注:Rg 与分子量的 1/3 次方大致成正比。
聚合物/RNA/DNA 类似规律:折叠/紧密堆积时 Rg 小;伸展时 Rg 大。
与 RMSD 的对比与互补
指标 | RMSD | Rg |
|---|---|---|
测量对象 | 与参考结构的差异 | 自身的紧凑程度 |
是否需要参考 | 需要(如晶体结构) | 不需要 |
反映信息 | 构象变化幅度 | 结构致密性 |
典型场景 | 是否回到天然态 | 是否发生膨胀/坍塌 |
联合解读
RMSD 高 + Rg 稳定 → 构象重排(如结构域相对转动),但整体仍紧凑
RMSD 高 + Rg 持续上升 → 蛋白质正在去折叠/变性
RMSD 低 + Rg 突然下降 → 可能发生局部坍塌或错误折叠成更紧但错误的结构
原子选择:
常用 Cα 原子(蛋白质)—— 简化计算且反映整体形状
全原子 Rg —— 更精确但受侧链摆动影响较大
对于配体/小分子,通常使用所有重原子
计算流程(以 GROMACS 为例):
gmx gyrate -s topol.tpr -f traj.xtc -o rg.xvg脚本/代码(Python + MDTraj):
import mdtraj as md
traj = md.load('traj.xtc', top='topol.pdb')
rg = md.compute_rg(traj)常见时间序列行为解读
时间曲线形态 | 含义 |
|---|---|
初始骤降后平稳 | 模拟开始时结构快速坍塌(常见于非天然起始结构) |
稳定值上下小幅波动 | 平衡态热波动,结构稳定 |
阶梯式跳跃 | 发生了离散的构象转变(折叠/去折叠事件) |
缓慢持续上升 | 逐渐变性/松散化(可能力场有问题或模拟时间不够) |
大幅度周期振荡 | 大尺度呼吸运动(某些柔性蛋白或核酸) |
第三个:SASA
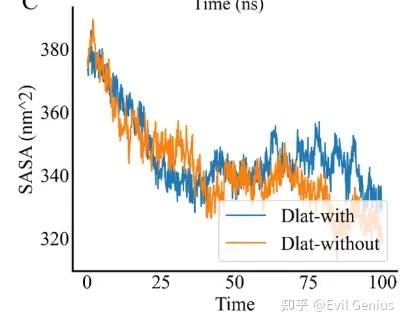
SASA(溶剂可及表面积)是分子动力学模拟中衡量分子与溶剂(通常是水)接触程度的关键几何参数。
溶剂可及表面积定义为:当一个半径为Rprobe的球形探针(代表水分子)在分子表面滚动时,探针中心所能到达的曲面所围成的面积。
常用探针半径:水分子通常取 1.4 Å(与水分子的范德华半径相关)。
直观理解:
SASA 值大 → 分子更多地暴露于溶剂 → 疏水区暴露、结构松散
SASA 值小 → 分子埋藏/被包裹 → 疏水核心致密、结构折叠
主要分析点
用途 | 说明 |
|---|---|
监测蛋白质折叠/去折叠 | 折叠时疏水残基埋入内部 → SASA ↓;去折叠时内部残基暴露 → SASA ↑ |
评估结构致密性与稳定性 | 平衡阶段 SASA 稳定波动 → 结构稳定;持续上升 → 变性/膨胀 |
表征疏水效应 | 疏水溶剂可及表面积(非极性 SASA)直接关联疏水相互作用的强度 |
分析配体结合 | 结合后界面区域的 SASA 减少(掩埋效应) |
判断残基埋藏/暴露 | 区分表面残基(高 SASA)与核心残基(低 SASA) |
验证模拟可靠性 | 与实验值(如通过化学位移或氘交换保护因子推导)对比 |
典型数值与含义
蛋白质(以小蛋白如溶菌酶~130残基为例):
状态 | SASA 相对变化 | 含义 |
|---|---|---|
完全折叠(天然态) | 最小(约 5000–7000 Ų) | 疏水核心形成,亲水表面暴露 |
部分去折叠 | 增加 20–50% | 内部残基开始接触溶剂 |
完全去折叠(无规卷曲) | 最大(增加 100% 以上) | 所有残基都暴露于溶剂 |
注:绝对 SASA 随分子量线性增长,通常比较相对变化(SASA(t) / SASA_native)。
与 Rg 的对比与互补
指标 | SASA | Rg(回转半径) |
|---|---|---|
物理含义 | 与水的接触面积 | 原子围绕质心的分布范围 |
反映信息 | 暴露/掩埋程度(化学环境) | 整体松散/紧凑程度(几何形状) |
对末端柔性的敏感度 | 中等(末端暴露,但总面积受整体影响) | 高(末端离质心远显著拉升 Rg) |
疏水效应表征 | 直接(可分解极性/非极性部分) | 间接 |
对局部膨胀的探测 | 灵敏(内部空洞暴露立即增加 SASA) | 相对不灵敏(空洞对质心分布影响小) |
联合解读:
Rg ↑ + SASA ↑ → 一致去折叠/膨胀(经典变性)
Rg ↑ + SASA 稳定 → 构象重排(如结构域相对分开,但接触面仍掩埋?需具体分析)→ 更常见:Rg ↑ 且 SASA 稳定少见,通常重排会改变暴露面积。更典型的是 Rg 稳定 + SASA 稳定 → 整体形状稳定;Rg 稳定 + SASA 小幅上升 → 内部空洞/裂缝增加但整体尺寸不变
Rg 稳定 + SASA ↑ → 分子内部出现空洞/裂缝或呼吸运动增加局部暴露(部分去折叠)
Rg ↓ + SASA ↓ → 一致坍塌压缩,可能过度折叠(或错误折叠成更紧但非天然的聚集体)
计算方法
算法:常用 Lee & Richards 的“滚动探针”算法或其变体(Shrake & Rupley、LCPO 等)。
残基/原子分解:
极性 SASA:N、O 及带电原子贡献 → 亲水相互作用
非极性 SASA:C、S 及芳香碳贡献 → 疏水效应(通常更关注变化)
计算流程:
GROMACS:gmx sasa(需分组文件 .ndx)
gmx sasa -f traj.xtc -s topol.tpr -o total_sasa.xvg -or residue_sasa.xvgAMBER (cpptraj):sasa 命令,可输出总 SASA、残基 SASA 及极性/非极性分解
VMD:插件 "SASA" 或 Tcl 命令 measure sasa
Python (MDTraj):mdtraj.shrake_rupley(traj, probe_radius=0.14)(注意单位:nm)
常见时间序列行为解读
时间曲线形态 | 含义 |
|---|---|
初始快速下降后平稳 | 模拟开始时结构自发折叠/坍塌(多见于起始结构非天然) |
稳定值附近小幅波动 | 平衡态热振动,结构稳定折叠 |
突然阶跃上升 | 去折叠事件(疏水核心突然打开)或配体解离 |
缓慢持续上升 | 渐进变性/膨胀(或力场参数问题) |
周期性振荡(高幅) | 大尺度“呼吸”运动(某些酶的功能相关) |
局限性及注意事项
探针半径固定(1.4 Å):对较小分子(如配体)或离子可能不合适,需调整探针半径。
不能区分“分子间”与“分子内”掩埋:SASA 降低可能来自自身折叠,也可能来自与其他分子的聚集。需结合聚集分析(如径向分布函数、低聚物检测)。
忽略溶剂动态与具体位置:两个残基可能暴露相同面积,但一个是部分埋藏,一个是完全暴露但面积相同?通常残基 SASA 需要归一化(参考三肽模型的最大暴露面积)。
残基 SASA 的涨落较大:单个残基的 SASA 随时间波动剧烈,分析时需取时间平均或滑动平均。
典型分析组合
组合 | 用途 |
|---|---|
SASA + Rg | 区分“整体膨胀”与“局部暴露” |
SASA + RMSD | 当 RMSD 上升时判断是去折叠(SASA↑)还是刚性重排(SASA稳定) |
极性/非极性 SASA 分开 | 分析疏水效应驱动 vs. 亲水相互作用变化 |
残基 SASA 热图 | 定位去折叠起始区域或配体结合热点 |
SASA vs. 时间 + 误差条(分块平均) | 评估平衡时 SASA 的收敛性(需多个独立模拟) |
第四点:RMSF(均方根波动)是分子动力学模拟中衡量分子每个残基/原子在模拟过程中的柔性或运动幅度的核心指标。
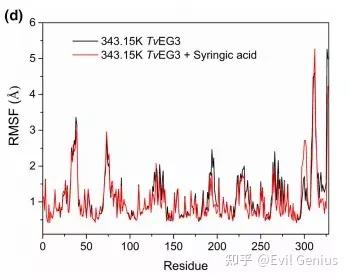
均方根波动计算的是每个原子(或残基)相对于其平均位置的波动大小的均方根值:
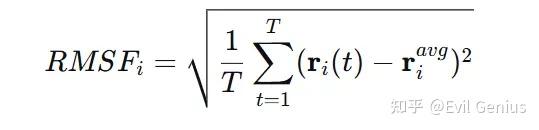
其中

主要分析
用途 | 说明 |
|---|---|
识别柔性区域 | 高RMSF → 灵活的运动区域(如 loop、末端) |
定位刚性区域 | 低RMSF → 结构保守区(如α螺旋核心、活性位点) |
评估模拟稳定性 | 整体RMSF低 → 结构稳定;过高 → 可能去折叠 |
验证实验数据 | 与NMR(核磁共振)的B-factor(温度因子)对比(B-factor ≈ 8π² × RMSF²) |
功能位点预测 | 活性位点/结合口袋通常刚性较低(或适度柔性) |
分析配体影响 | 结合配体后 → 结合位点周围RMSF降低(刚性增强) |
蛋白质的典型RMSF分布:
区域 | RMSF | 相对值 示例 |
|---|---|---|
α-螺旋核心 | 低 | 0.5-1.5 Å |
β-折叠片 | 中低 | 1-2 Å |
loop/转角 | 中高 | 2-4 Å |
N-/C-末端 | 最高 | 3-8+ Å |
配体结合位点 | 低(结合后) | 0.5-1.5 Å |
理想情况:核心区RMSF < 2 Å;末端< 5 Å;无大幅spike(跳跃)。
与RMSD的对比互补
指标 | RMSD | RMSF |
|---|---|---|
对象 | 整个分子的整体差异 | 每个残基的局部波动 |
时间性 | 每个时刻 vs. 参考结构 | 对整个时间轨迹统计 |
揭示信息 | 是否接近参考构象 | 哪些区域更灵活/刚性 |
发现稳定性 | 轨迹整体是否收敛 | 哪些部分主导运动 |
联合解读:
RMSD稳定 + RMSF低 → 理想稳定模拟,刚性折叠
RMSD稳定 + RMSF局部高 → 柔性区域(如酶的功能loop)不影响整体折叠
RMSD上升 + RMSF普遍升高 → 全局去折叠/变性
RMSD上升 + RMSF仅末端升高 → 末端呼吸运动,核心仍稳定
计算要点
原子选择:
通常用 Cα 原子(蛋白主链)—— 代表骨架柔性
全原子RMSF → 侧链柔性更大,反映局部化学环境
可自定义分组(如结合位点残基)
计算流程:
GROMACS:gmx rmsf
gmx rmsf -f traj.xtc -s topol.tpr -o rmsf.xvg -res # 残基级别AMBER (cpptraj):atomicfluct 或 rmsf 命令
VMD:measure rmsf(需先使用 measure fit 对齐)
Python (MDTraj):
import mdtraj as md
traj = md.load('traj.xtc', top='topol.pdb')
rmsf = md.rmsf(traj, traj, frame=0) # 相对平均结构的RMSF重要步骤:
对齐(拟合):计算前必须先将轨迹与参考结构(如平均结构)对齐,否则整体平动/转动会被误算为波动。
去除平动/转动:常用最小二乘拟合(如拟合到Cα骨架)。
可视化细节
残基序号 vs. RMSF:柱状图/线图,用颜色标记二级结构(螺旋=红色/折线,折叠=蓝色/箭头,loop=灰色)
结构着色(PyMOL/VMD):
蓝色 → 刚性(低RMSF)
白色 → 中等
红色 → 柔性(高RMSF)
热图:时间×残基的波动矩阵(识别协同运动)
注意事项
不能区分运动类型:高RMSF可能是随机热振动,也可能是协调的功能运动(如开合)。需结合动态相关矩阵或主成分分析。
对末端过度估计:蛋白末端天然游离,RMSF很高但不代表功能重要性。可排除末端(如前5/后5残基)再分析。
依赖对齐区域:如果整体结构域相对运动,使用全蛋白对齐会放大特定区域的RMSF。可考虑分结构域对齐后再计算。
绝对值受模拟时长影响:短模拟可能低估RMSF(未充分采样)。需检查收敛性(计算不同时间窗口的RMSF)。
实际运用
设计抑制剂后,评估靶蛋白稳定性
野生型:活性位点 RMSF = 1.8 Å(适度柔性)
突变型:活性位点 RMSF = 3.5 Å(柔性增加)→ 可能丧失功能
加抑制剂:活性位点 RMSF = 0.9 Å(刚性增加)→ 结合成功稳定位点
第五部分:氢键分析是分子动力学模拟中用于研究分子间或分子内特定相互作用的重要方法,尤其在蛋白质、核酸及蛋白质-配体复合物体系中。
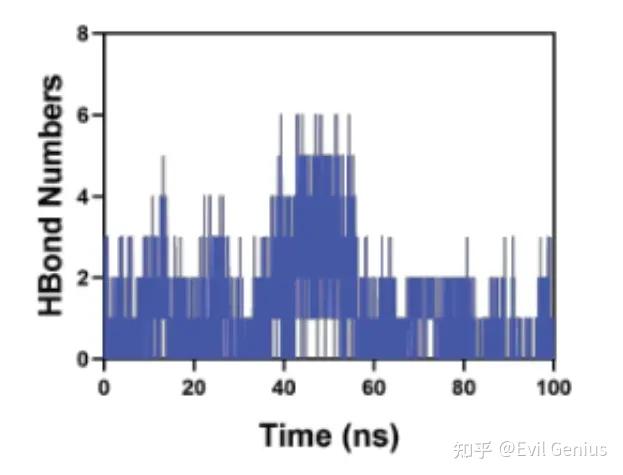
氢键的定义与几何判据
在MD模拟中,一个氢键通常被认为成立,当同时满足以下两个几何条件:
距离准则:氢键受体 (A) 与供体 (D) 上的极性氢 (H) 之间的距离小于某阈值,通常为 3.5 Å(或 3.0-3.5 Å,取决于定义)。
角度准则:供体-氢-受体 (D-H···A) 形成的角度大于某阈值,通常为 120° 或 150°(角度越接近 180°,氢键越强)。
注:更精确的模型(如用于水或离子液体的)可能使用能量或拓扑准则,但在生物大分子模拟中,几何判据因其计算效率高而占据绝对主导。
主要用途
用途 | 说明 |
|---|---|
评估结构稳定性 | α-螺旋、β-折叠中规则的主链内氢键数量多→ 二级结构稳定 |
检测蛋白质折叠/去折叠 | 去折叠过程中,二级结构氢键大量断裂 → 氢键总数锐减 |
分析配体结合模式 | 配体与靶点间关键氢键在模拟中持续存在 → 结合可靠 |
研究溶剂效应 | 蛋白质-水氢键 → 水合作用;水-水氢键 → 有序水分子 |
比较突变影响 | 关键残基突变 → 周围氢键网络改变 → 功能变化 |
解释功能运动 | 别构效应常通过破坏或形成长程氢键网络实现 |
常见分析类型
(1) 氢键数量随时间变化
分子内氢键(蛋白):总氢键数在平衡区稳定 → 结构稳定;突然下降 → 结构崩溃或去折叠。
分子间氢键(蛋白-配体):高且持续 → 强结合;波动大 → 弱、非特异性结合。
蛋白-水氢键:反映水合层结构与动力学。
(2) 氢键占用率(Occupancy)
定义为氢键存在的时间比例(帧数/总帧数)× 100%。
> 90%:非常稳定,关键结构/结合氢键
50–90%:中等稳定,动态断裂-形成
< 50%:瞬时/弱相互作用,可能仅是偶发水桥接
0%:从未形成
(3) 氢键寿命与动力学
连续寿命:氢键从形成到断裂的连续存在时间。
间歇寿命:考虑断裂后又重新形成的总关联时间(包括中间断裂)。
水分子与蛋白之间的氢键寿命通常很短(数ps),而二级结构中主链氢键可达ns级。
(4) 氢键网络与聚类
分析水分子或侧链形成的氢键网络、环或桥接结构,常用于研究质子传递、水通道等。
计算方法与工具
工具 | 典型命令/函数 |
|---|---|
GROMACS | gmx hbond(需索引文件指定供体/受体组) |
AMBER (cpptraj) | hbond 命令,可自动识别或自定义 |
VMD | 插件 “Hydrogen Bonds” 或 Tcl 循环 hbonds |
MDTraj (Python) | mdtraj.kabsch_sander(对蛋白二级结构氢键专用)或 mdtraj.wernet_nilsson(水分子) |
LIGPLOT | 仅静态结构,需配合MD取平均结构或代表性帧 |
GROMACS 蛋白-配体氢键分析
# 生成索引(假设配体为组 LIG)
gmx make_ndx -f complex.gro -o index.ndx
> del 0-2 # 删除默认组
> ri 1-100 # 选择受体蛋白
> ri 101 # 选择配体
> name 1 Protein
> name 2 Ligand
> q
# 分析氢键(默认距离3.5Å,角度30°)
gmx hbond -f traj.xtc -s topol.tpr -n index.ndx -num hb_num.xvg -hbn hb_network.ndx -hbm hb_matrix.xpm -life hb_life.xvg结果解读与典型模式
蛋白质内部氢键
状态 | 特征 |
|---|---|
天然折叠 | 主链氢键数高且稳定(约 0.7-0.9 氢键/残基,二级结构区占用率 > 90%) |
部分去折叠中间态 | 氢键数下降 20–40%,波动增大 |
完全去折叠 | 主链氢键数很少,残基之间无规则 |
蛋白-配体氢键
占用率范围 | 含义 |
|---|---|
90–100% | 强、稳定的特异性相互作用,是结合的关键锚点 |
60–90% | 重要贡献,结合袋内动态但持续相互作用 |
30–60% | 辅助结合,可能与水介导氢键竞争 |
< 30% | 瞬时或非特异性,不太可能是主要驱动力 |
溶剂氢键
蛋白-水:表面亲水残基(如 Arg、Lys、Glu、Asp)和主链羰基氧是常见的蛋白质-水氢键受体。
水-水:第一水合层内水分子之间氢键网络的扭曲程度反映表面化学性质。
注意事项
判据选择:角度阈值 120° vs. 150° 可显著影响占用率报告。同一研究中保持参数一致,与文献对比时注意参数差异。
氢原子的位置:经典力场(如 Amber、CHARMM)使用隐式氢模型(非极性氢合并到碳上),此时只分析“可形成氢键的极性氢”(连接到 N、O 的氢)。需确保拓扑文件中极性氢存在且位置合理。
水介导的氢键:直接 hbond 分析默认忽略水桥。可使用“水桥分析”(gmx hbond -water)识别配体-水-蛋白间接氢键。
虚假氢键:当供体、受体间距离小时但角度很差(例如 < 90°)时几何判据会误判。可用更严格的能量判据或手动验证关键氢键。
统计收敛:氢键占用率需要足够长的模拟时间(至少几十 ns 至几百 ns)才能收敛。监测占用率随时间窗口长度的变化判断收敛性。
与其它分析指标的联合解读
组合 | 目的 |
|---|---|
氢键 + RMSD | RMSD 稳定但关键氢键占用率低 → 可能接近天然态但结合位点不稳定 |
氢键 + RMSF | RMSF 高的 loop 区氢键占用率低 → 柔性由局部氢键断裂导致 |
氢键 + SASA | 疏水核心 SASA 增大 + 内部氢键断裂 → 去折叠 |
氢键 + 结合自由能分解(如 MM-GBSA) | 识别对结合贡献大的关键极性相互作用 |
第六点:Gibbs自由能(G)是判断一个过程(如结合、折叠、相变)能否自发进行的最核心热力学量。
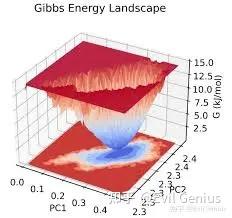
MD模拟天然给出的是势能和动能(总能量),而Gibbs自由能同时包含了熵的贡献G=H−TS),无法直接从轨迹的平均值中读取。因此,我们需要专门的“自由能计算方法”来得到它。
以下是MD中计算Gibbs自由能差ΔG)的主流方法:
方法 | 核心原理 | 精度 | 计算量 | 最佳适用场景 |
|---|---|---|---|---|
热力学积分 (TI) | 构建一个从状态A到B的非物理“炼金术”路径,对路径上的能量梯度进行积分。 | 非常高 (黄金标准) | 极大 (需模拟多个中间态) | 精确计算两个相似分子(如药物衍生物)的相对结合自由能。 |
自由能微扰 (FEP) | 与TI类似,但基于Zwanzig方程,通过计算状态间的能量差来获得自由能变化。 | 非常高 (黄金标准) | 极大 (需充分的相空间重叠) | 与TI应用场景类似,是药物设计中高精度计算的首选之一。 |
MM/PB(GB)SA | 对MD轨迹中的大量快照,用连续介质模型近似溶剂,计算平均能量和熵变。 | 中等 (依赖力场和采样) | 低 (后处理分析) | 快速估算蛋白质-配体结合自由能,或在大规模虚拟筛选中排序候选分子。 |
伞形采样 (US) | 沿反应坐标(如距离、角度)施加偏置势,迫使体系跨越能垒,再通过WHAM/MBAR重构平均力势(PMF)。 | 中等 (高依赖反应坐标选择) | 中等 (需并行窗口采样) | 研究解离、构象变化等动力学过程的自由能曲线(能垒)。 |
界面钉扎 (IP) | 施加约束迫使体系保持两相共存(如固+液),测量维持该状态所需的力,从而推导出Gibbs自由能差。 | 高 | 中等 | 计算固-液相变(如冰的熔点)、材料科学中的相图。 |
方法选择指南
- 如果是药物筛选/突变效应:首选 MM/PB(GB)SA(快速)或 FEP/TI(高精度,仅限小规模关键分子)。
- 如果研究配体解离或构象变化:首选 伞形采样 (US) 来计算PMF。
- 如果研究材料相变(如凝固点):可尝试 界面钉扎 (IP) 或自由能微扰
核心难点与现状
- 熵的精确计算极其困难:虽然通过谐振近似(NMA)或准谐波分析(QHA)可估算熵,但会忽略蛋白质真正的非谐波运动(如侧链翻转、loop跳跃),导致误差较大。
- 计算与精度权衡:
- FEP/TI:误差通常在 1-2 kcal/mol(化学精度),但耗时巨大。
- MM/PBSA:相关误差可能在 2-4 kcal/mol,但速度快上百倍,适合排序。
- 经验法则:如果配体结构差异>10个重原子,FEP/TI路径可能过于复杂,此时MM/PBSA或LIE可能更实用。
第七点:配体的径向分布函数(Radial Distribution Function, RDF) 是分子动力学模拟中研究配体分子与目标蛋白之间空间分布和结合模式的关键工具。
它回答了一个核心问题:配体分子更倾向于待在哪里?
径向分布函数 g(r)?
径向分布函数 g(r)是一个统计量,它量化了在距离一个参考粒子 r处,找到另一个粒子的概率密度与完全随机分布(平均密度)的比值
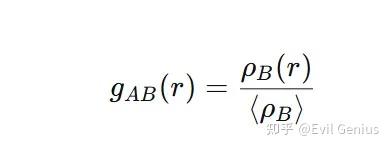

直观理解:
g(r)=1:在该距离上,B粒子的分布是随机的,与背景密度无差异。
g(r)>1:在该距离上,B粒子的出现概率高于平均值,表明两者之间有吸引相互作用或特定的堆积模式。
g(r)=0:在该距离上,B粒子从未出现,通常是因为粒子间的强排斥作用(空间位阻或静电排斥)。
在蛋白质-配体模拟中,我们通常计算配体的某个关键原子(或其质心)相对于蛋白质上特定原子(或质心)的 g(r)。
配体RDF分析的核心目的
与连续相(如溶剂水)的RDF最终会收敛到1不同,配体作为离散的“溶质”,其RDF分析有更具体的应用场景:
分析目标 | 说明 |
|---|---|
确定结合模式与距离 | 通过观察RDF的第一主峰位置,可以精确量化配体是否紧密地结合在目标口袋中,及其关键官能团与蛋白残基的距离。 |
评估结合稳定性 | 峰形尖锐且高度高,表明配体在口袋中的位置和取向非常稳定、明确;峰形宽而矮,则表示配体在结合位点内或周围有较大的柔性运动。 |
识别特定的相互作用 | 通过分别计算配体上的疏水基团/极性基团与蛋白上特定残基的RDF,可以推断出具体的疏水相互作用或氢键网络。 |
验证或反驳结合模式 | 将从模拟轨迹计算出的RDF特征(如特征峰位)与对接预测的模型或实验结果(如NMR距离约束)进行对比,验证模拟的可靠性。 |
研究非经典结合 | 识别配体是否在蛋白表面的多个“热点”区域之间跳动,或发现意料之外的变构结合位点。 |
计算方法与工具
计算的核心在于合理定义“参考组”和“目标组”。
- 参考组:通常可以是蛋白质的结合口袋、特定残基、蛋白质的质心,甚至是蛋白质的整个表面。
- 目标组:可以是整个配体分子的质心,或者是配体上的某个特定原子(如疏水尾巴的末端碳、氢键供体/受体原子)。
示例:GROMACS 计算命令
GROMACS提供了非常方便的工具 gmx rdf。例如,以下命令计算了在模拟的最后15 ns内,配体(索引组 1ZIN)相对于蛋白质表面(-surf mol)的径向分布:
gmx rdf -s prolig.tpr -f prolig_fit.xtc -o rdf_1_surf.xvg \
-n ZIN.ndx -ref protein -sel 1ZIN -bin 0.01 -surf mol \
-b 85000 -e 100000 -pbc yes-ref protein:计算以蛋白质为参考。
-sel 1ZIN:计算配体的分布。
-surf mol:这是一个关键选项,告诉程序计算配体到蛋白质表面(而非到蛋白质的某个原子)的距离,这能更好地处理凹凸不平的蛋白质表面。
-b 和 -e:定义了分析的起始和结束时间。
生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号