AlphaFold 折叠蛋白,LiteFold 折叠数据 | LiteFold 扔出 TB 级蛋白数据

AlphaFold 折叠蛋白,LiteFold 折叠数据 | LiteFold 扔出 TB 级蛋白数据

MindDance
发布于 2026-05-29 13:14:13
发布于 2026-05-29 13:14:13

过去几年,蛋白质 AI 的变化很容易被一句话概括:模型越来越强了。
AlphaFold 让结构预测从少数结构生物学团队的专门能力,变成很多实验室都能调用的基础工具。ESM 这类蛋白语言模型,把序列、进化信息和结构表征推到了更大的尺度。蛋白设计、突变效应预测、稳定性建模、抗体与多肽工程,也都开始把深度学习模型放进工作流里。
但真正开始训练模型、复现实验、搭建 benchmark 时,问题往往不是模型先卡住,而是数据先卡住。
蛋白质数据不是一张干净的表。它可能藏在论文补充表里,挂在 FTP 目录中,分散在 UniProt、PDB、AlphaFold DB、ESM Atlas、ProteinGym、MegaScale、Pfam、InterPro、GOA 等不同来源里。一个数据源按序列组织,另一个按结构链组织;一个分数越高越好,另一个分数越低越稳定;一个记录突变效应,另一个记录结构坐标;一个有许可说明,另一个需要追溯上游版本。
所以,当 LiteFold 发布 AminoWeb 时,它真正切入的并不是一个新的蛋白质模型,而是一个更底层的问题:
如果蛋白质 AI 已经进入大模型时代,支撑它的数据层是否也该变得更像机器学习基础设施?

它到底在做什么?
AminoWeb 是 LiteFold 在 Hugging Face 上发布的一组蛋白质机器学习数据集合。按照发布信息,它包含 29 个经过清洗、面向机器学习整理的数据集,总体量约 7.56 TB;Hugging Face collection 页面则将其概括为约 8 TB 的蛋白质数据集合。
它覆盖的不是单一任务,而是一组蛋白质机器学习常见输入与监督信号:序列、结构、功能注释、多序列比对、突变效应、稳定性、结合与相互作用等。
换句话说,它不是在提出一个新的结构预测模型,也不是给出一个新的药物设计 pipeline。它更像是把蛋白质 AI 里常见的数据源,重新整理成模型工程师更容易读取、检索、训练和复现实验的形态。
这件事看起来不如新模型显眼,但它解决的是另一个更日常的问题:数据能不能被稳定地读进来,能不能知道每一行从哪里来,能不能避免训练和测试之间因为同源蛋白泄漏而虚高,能不能在不同任务之间复用。
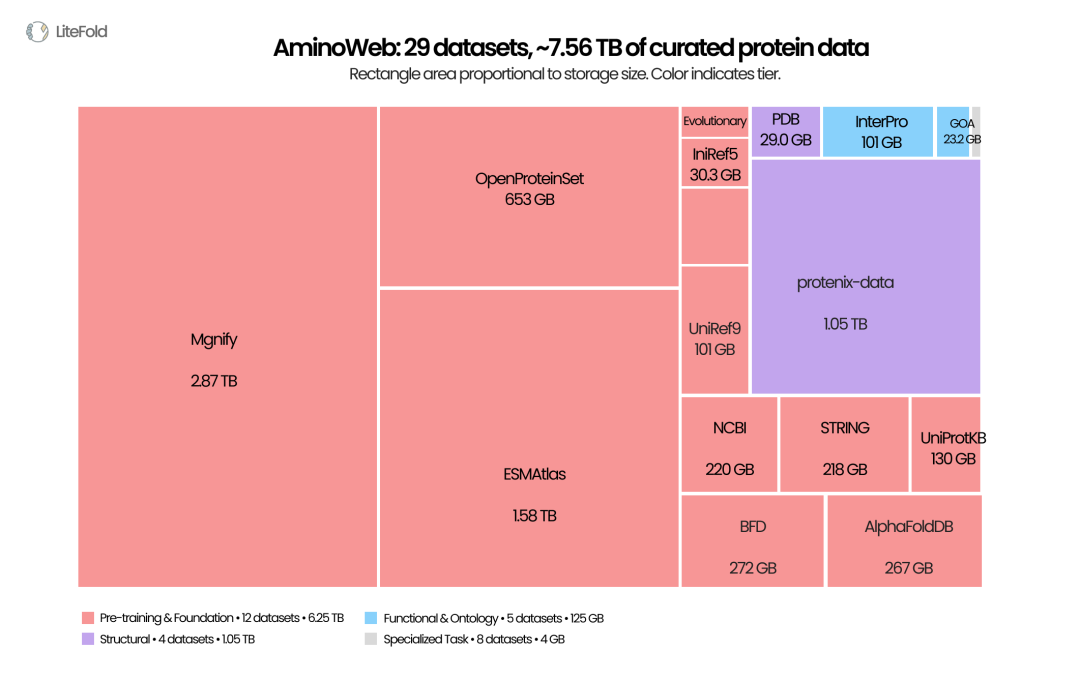
AminoWeb 将序列、结构、功能、突变效应、稳定性和相互作用等蛋白质数据整理为可访问的数据集合。图中面积反映数据体量,不代表数据质量或任务重要性
AminoWeb 将序列、结构、功能、突变效应、稳定性和相互作用等蛋白质数据整理为可访问的数据集合。图中面积反映数据体量,不代表数据质量或任务重要性
问题为什么会出在数据层?
大语言模型领域早就习惯了把数据集当成基础设施来讨论。FineWeb、The Pile、RedPajama、Dolma 这些名字,本质上都不只是数据本身,也是训练语料如何收集、清洗、去重、记录来源和服务模型训练的工程实践。
蛋白质机器学习的问题更复杂。
自然语言里,一段网页文本和另一段网页文本之间当然也会有重复和污染,但蛋白质数据里有一种更隐蔽的重复:同源性。两个蛋白序列可能不完全相同,却来自共同祖先,结构和功能高度相关。如果训练集和测试集里出现高度相似的同源蛋白,模型看起来可能泛化很好,实际却只是学到了相近家族的模式。
这也是蛋白质 benchmark 里经常让人头疼的地方。随机划分很方便,但随机划分未必能模拟真实发现任务。真正困难的场景,往往是让模型面对远离训练分布的新家族、新结构域、新突变组合,甚至是实验注释很少的序列空间。
还有一个问题是分数口径。
突变效应数据、稳定性数据、结合数据、功能注释数据,看起来都可以变成监督学习任务,但它们的分数含义并不相同。有的分数表示活性升高,有的表示适应度下降,有的表示 ΔΔG,有的来自高通量实验,有的来自人工整理数据库。如果为了统一格式而粗暴统一方向,反而可能把原始生物学含义洗掉。
所以,蛋白质数据整理不是简单地把 CSV 转成 Parquet。更关键的是,保留原始口径,同时让模型工程可以稳定使用。
AminoWeb 把哪些东西放到了一起?
从 collection 页面看,AminoWeb 覆盖了多类常用蛋白质数据源。它们大致可以分成几组。
数据类型 | 代表数据源 | 对模型训练有什么用 | 需要注意什么 |
|---|---|---|---|
序列与同源信息 | UniProtKB、UniRef50、UniRef90、BFD、MGnify、NCBI | 训练蛋白语言模型,构建 MSA,做序列检索与家族分析 | 序列冗余很高,划分时要控制同源泄漏 |
结构数据 | PDB、AlphaFoldDB、ESMAtlas、CATH | 结构预测、结构表征学习、结构检索、结构条件设计 | PDB 是实验结构,AlphaFoldDB 和 ESMAtlas 是预测结构,不能混为一谈 |
功能与注释 | GO、GOA、InterPro、Pfam、Human Protein Atlas | 功能预测、结构域识别、蛋白家族建模、多任务学习 | 注释密度不均,热门蛋白和模式物种会被过度代表 |
突变效应与稳定性 | ProteinGym、MegaScale、FireProtDB、FLIP2 | 突变效应预测、蛋白稳定性建模、序列优化 | 不同实验体系的分数方向和噪声水平不同 |
结合与相互作用 | SKEMPI2、IEDB、STRING | 蛋白互作、抗原表位、结合变化预测 | 数据来源混合,实验置信度与计算预测需要区分 |
化学与特殊残基 | PDB-CCD、SwissSidechain、CycPepMPDB | 处理配体、非天然氨基酸、环肽等非标准对象 | 对药物发现很重要,但标准蛋白模型未必天然支持 |
为了方便读者理解,将 collection 中可见的数据源按常见蛋白质机器学习任务重新归类。
这张表能说明 AminoWeb 的定位。
它试图把蛋白质 AI 的几个关键数据面放到同一入口下:序列空间、结构空间、功能注释、实验测量、相互作用和非标准化学组件。
对于做模型的人来说,这意味着一件很实际的事:不必每次都从不同数据库重新写下载脚本、解析脚本和字段映射脚本。至少在起步阶段,可以先用一个统一的数据入口搭建任务。
旧方案为什么不够?
过去做蛋白质机器学习,经常是每篇论文自己整理一份数据。
这在小规模研究里不是问题。研究者可以下载一个补充表,写脚本清理突变格式,再按照论文定义划分训练集和测试集。问题出现在模型和任务规模变大以后。
第一,清洗过程很难复现。
同一个数据源,是否去掉缺失值,是否合并重复突变,是否保留低置信度结构,是否统一 isoform,是否筛掉过长序列,都会影响结果。论文里通常不会把每一步工程细节写到足够细。
第二,跨任务复用很困难。
一个模型可能想同时用序列、结构、功能、突变效应和稳定性数据。每个数据源都有自己的主键和字段习惯。UniProt accession、PDB chain、AlphaFold ID、MGnify ID、突变 notation、结构域坐标,如果没有统一映射,后续建模会消耗大量时间。
第三,评测容易被数据划分影响。
蛋白质任务尤其怕训练测试之间存在高度相似的同源蛋白。一个模型在随机划分上表现很好,不一定意味着它真的学会了远距离泛化。很多时候,benchmark 的可信度取决于数据划分,而不只取决于模型指标。
这也是 AminoWeb 提到 homology-aware splits 的原因。它指向的不是格式问题,而是评测可信度问题。
新设计解决了什么?
AminoWeb 的核心价值可以概括为四层。
第一层是格式。
数据被整理成更适合机器学习读取的形式,例如 Parquet、JSONL、WebDataset 等。Parquet 对大表、列式读取和类型约束更友好;JSONL 保留原始行信息比较方便;WebDataset 更适合承载大规模结构文件或分片数据。
这一步看似工程化,却会直接影响使用门槛。对于 TB 级数据,能不能 streaming,能不能只读部分字段,能不能按 shard 下载,差别很大。
第二层是来源追踪。
在多个 dataset card 中,可以看到类似 dataset_id、source_file、row_index、raw upstream row 这类设计。它们的意义是:模型读到一行数据时,理论上可以追溯它来自哪个上游文件、哪一行、原始字段是什么。
这对科学数据很重要。因为蛋白质数据库不是静态对象,版本会更新,字段会变化,注释会修订。如果没有 provenance,模型结果出了问题,很难追到源头。
第三层是分数口径。
AminoWeb 发布信息中特别提到 preserved score conventions,也就是保留原始分数约定。这个决定很重要。
很多人做数据集整理时,会倾向于把所有分数都转成统一方向,例如越大越好。但在蛋白质实验数据里,这样做可能会抹掉上游实验语境。更稳妥的做法是保留原始分数,同时在 dataset card 或字段说明里告诉用户如何解释。
第四层是评测划分。
蛋白质任务的划分不能只看样本随机性,还要看序列、结构和家族关系。AminoWeb 将 homology-aware splits 作为发布亮点之一,说明它意识到蛋白质 ML 的核心风险之一是同源泄漏。
不过这里也要克制理解。不是所有子数据集都天然拥有同一种划分方式,具体到某个任务,仍然需要查看对应 dataset card,确认划分规则、字段含义和适用边界。
结果到底说明了什么?
从这次发布能看到几个信号。
AlphaFoldDB 在 AminoWeb 中被整理为 prediction index,数据页显示约 2.47 亿行,并划分为 train 与 test。这里要注意,它更像是预测结构索引和元数据表,而不是简单等同于完整结构坐标仓库。
ESMAtlas 数据页显示为大规模 WebDataset 镜像,文件体量约 1.74 TB,并保留结构文件、metadata、confidence scores 等信息。它还明确提醒:ESMAtlas 结构是计算预测,不是实验结构,下游任务需要用 pLDDT、pTM 等置信度指标筛选。
ProteinGym 数据页显示约 293 万行,面向蛋白适应度和突变效应预测,是评估蛋白语言模型、逆折叠模型和监督预测模型的重要 benchmark。
MegaScale-Tsuboyama2023 数据页显示约 29.9 万行整理记录,对应大规模蛋白折叠稳定性实验数据。它的价值在于把结构之外的热力学稳定性信号带入模型训练和评估。
Pfam 数据页显示约 1.28 亿行,主要承载蛋白家族和结构域区域注释。对于功能预测、结构域识别和蛋白家族学习,这类注释数据比单纯序列更接近可解释的生物学标签。
这些数字不该被理解为越大越好。更准确的理解是:AminoWeb 把不同粒度的蛋白质数据放到了一个相对统一的机器学习入口下,让研究者能够更快构造跨数据源任务。
但它也不能解决所有问题。
预测结构仍然是预测结构。低置信度区域、无序区、复合物界面、构象变化和活性位点几何,仍然需要谨慎使用。功能注释仍然存在偏倚。突变效应数据仍然受实验体系影响。稳定性数据和结合数据也不能直接互相替代。
数据被整理好,并不等于生物学问题被解决。
对蛋白质 AI 来说,它意味着什么?
AminoWeb 的意义不在于它让某个模型立刻变强,而在于它把蛋白质 AI 的一个长期瓶颈显性化了:模型规模扩张以后,数据层也必须进入工程化阶段。
这会影响几类工作。
对基础模型训练来说,统一的数据入口可以降低预训练、微调和多任务学习的工程成本。序列、结构、MSA、功能注释、突变效应和稳定性数据如果更容易组合,模型就更容易从单一任务走向多模态蛋白质表征。
对 benchmark 来说,来源追踪和划分规则会变得更重要。未来评价蛋白质模型,不能只看指标,还要看测试集和训练集之间的同源关系、数据版本、标签来源和任务定义。
对药物发现和蛋白工程来说,AminoWeb 这类数据集合可能会让更多团队快速搭建原型。例如先用 ProteinGym 做突变效应评估,再结合结构数据和稳定性数据筛选候选,再把相互作用或表位数据接入后续分析。它不会替代实验,但可以让计算筛选更可复现。
对开源生态来说,这也可能推动蛋白质 AI 从论文数据走向数据资产。一个数据集不只是下载链接,而是要有字段、版本、许可、来源、拆分、使用示例和限制说明。
这件事听起来不像模型发布那样兴奋,但它可能更接近领域真正需要的基础设施。
参考文献
[1]. LiteFold. AminoWeb Collection. https://huggingface.co/collections/LiteFold/aminoweb
[2]. LiteFold
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-29,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号