CosMx文献分享--结直肠癌中KRAS抑制剂并发性遗传与非遗传耐药机制
原创
CosMx文献分享--结直肠癌中KRAS抑制剂并发性遗传与非遗传耐药机制
原创
追风少年i
发布于 2026-06-02 09:40:39
发布于 2026-06-02 09:40:39
作者,Evil Genius
大家不要以为基础分析很简单,大家先看下面
技术平台 | 样本类型 | 推荐切片厚度 |
|---|---|---|
Xenium | FFPE (石蜡切片) | 5 µm |
Fresh Frozen (新鲜冷冻) | 10 µm | |
Visium / Visium HD | FFPE | 5 µm |
Fresh Frozen | 10 µm | |
Stereo-seq | FFPE | 5 µm |
Fresh Frozen | 10 µm | |
CosMx | FFPE | 5 µm |
Fresh Frozen | 5 µm (可切至10µm,但仪器仅采集最表面5µm) |
首先回答一个问题,常见的哺乳动物的细胞直径是多少(看过B站课程或者参加过培训的应该知道)?5um和10um厚度的切片,质控标准一样么?低精度visium都说基因表达下限是500,冰冻切片还是FFPE?如果冰冻切片是500,FFPE的应该调整为多少?
今时不同往日了,科研来讲,很多时候都是细节决定成败。
今天我们分享文献
知识积累
结直肠癌是全球癌症死亡的第二大原因,KRAS突变存在于近50%的患者中。
尽管KRAS抑制剂(如adagrasib、sotorasib)已获批用于治疗,但仅少数结直肠癌患者有效,且中位无进展生存期不足7个月。
耐药机制不完全清楚:部分患者出现新的基因突变(cfDNA中可检测),但往往丰度低、呈亚克隆性;部分患者则完全没有明确的遗传耐药机制。
核心发现
遗传与非遗传耐药并存:同一患者体内,既有亚克隆性的获得性基因突变,也存在转录水平的适应性改变(非遗传机制)。
肿瘤内高度异质性:不同肿瘤区域的耐药状态不同,间充质、YAP、胎儿样转录特征占主导,而炎症程序在治疗早期即被激活。
炎症程序是癌细胞自主性的:通过类器官模型证实,药物诱导的炎症反应先于耐药出现,且至少部分由癌细胞自身驱动。
潜在治疗策略:识别出 TBK1 作为靶点,联合KRAS抑制剂与TBK1抑制剂,有望阻断早期炎症适应阶段,增强治疗反应。
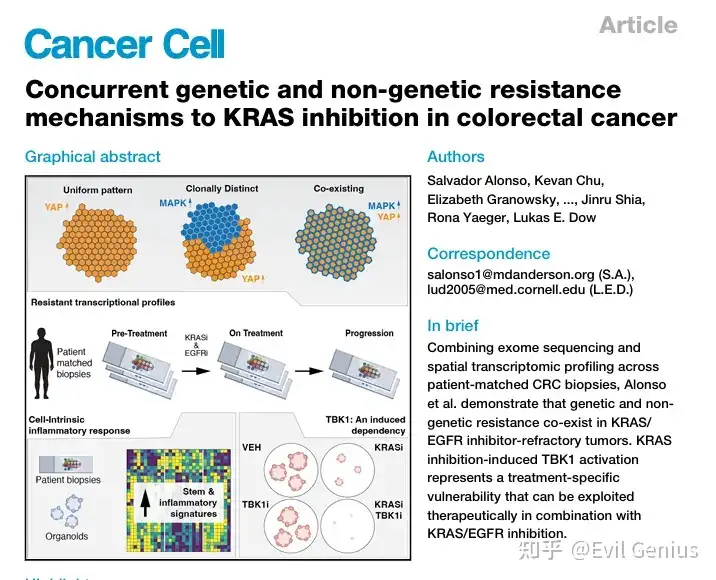
结果1、结直肠癌(CRC)对KRAS抑制剂产生遗传与非遗传适应性
从12名接受KRAS612C抑制剂联合EGFR抑制剂治疗的CRC患者中,前瞻性收集了治疗前(11例)、治疗中(第7-21天,11例)和耐药后(8例) 的活检样本。
治疗方案包括索托拉西布+帕尼单抗,或阿达格拉西布±西妥昔单抗。
患者既往均接受过化疗;5例部分缓解,7例疾病稳定;平均治疗持续9.4个月。
遗传耐药机制(靶向外显子测序):
在11例有配对样本的患者中,7例发现了获得性遗传事件,包括KRAS⁶¹²C扩增、FLT3扩增,以及EGFR、FLT3、DUSP4、ROS1的错义突变。
大多数耐药驱动突变呈亚克隆性(17个新生突变中仅7个为克隆性突变)。
其余4例患者未检测到明确的获得性遗传事件 → 提示非遗传机制也参与耐药。
非遗传/表型适应(空间转录组学,CosMx平台):
分析了近68万个细胞(其中约37.6万个癌细胞),发现:
癌细胞按患者个体和治疗状态聚集;
治疗前和耐药后样本的转录组异质性较高,而治疗中样本异质性降低、聚集更紧密;
肿瘤内转录异质性在不同治疗阶段相似。
细胞组成变化:
治疗早期癌细胞数量显著减少;
癌症相关成纤维细胞、B细胞、内皮细胞比例增加(B细胞变化在排除癌细胞后仍有统计学意义);
通过多重免疫荧光补充分析发现,治疗样本中CD4+ T细胞显著富集(CosMx未很好捕获该群体),其他免疫/基质细胞无明显差异。
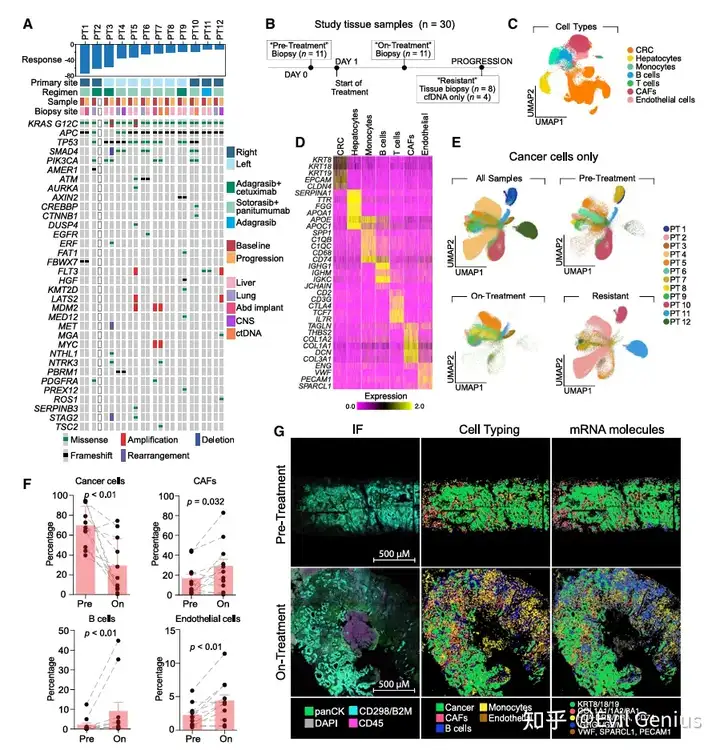
KRAS抑制剂治疗过程中,结直肠癌细胞的转录适应性变化
分析恶性细胞(癌细胞) 在治疗前、治疗中、耐药后的差异表达基因与信号通路。
使用功能富集分析(ORA)和基因集变异分析(GSVA),覆盖经典Hallmark通路及肠道细胞类型特异性基因集。
治疗中(on-treatment)的适应性特征:
出现肠道潘氏细胞和干细胞特征激活。
显著富集干扰素(IFN)和细胞因子信号通路等炎症程序。
肠上皮分化和肠分化标志物明显下调,而肠道干细胞标志物上调。
KRAS靶基因表达水平较治疗前降低。
耐药后(resistant)的适应性特征:
富集上皮-间充质转化(EMT)、胎儿样程序和YAP信号通路——这些特征已知与结直肠癌转移及WNT靶向治疗耐药相关。
肠上皮分化标志物的下调在多数患者中恢复至基线水平。
KRAS靶基因表达在部分患者中再次升高,可能与停药/治疗压力改变有关。
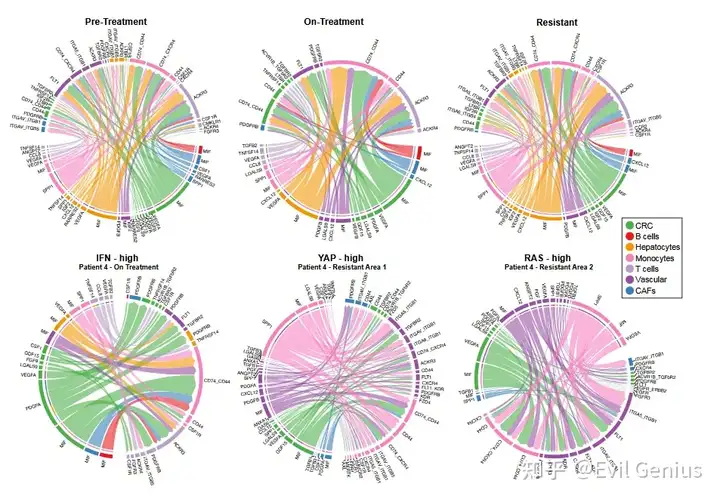
关键例外个案(患者4):
与其他患者不同,该患者在治疗前即表现出强烈的I型和II型干扰素程序表达。
该患者因类风湿关节炎发作,在治疗前及治疗中分别使用了羟氯喹(抗炎)和利妥昔单抗,可能影响了基线转录状态。
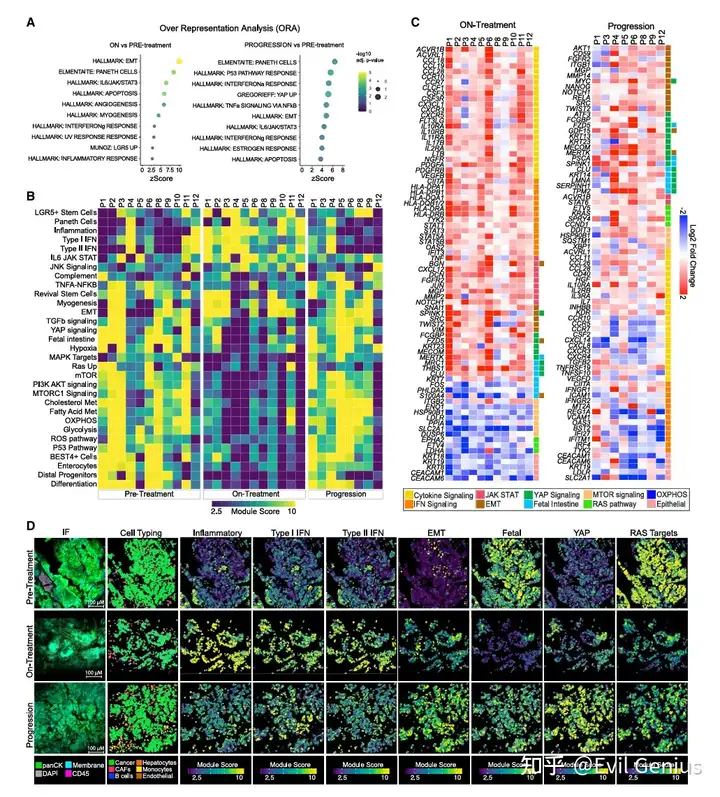
结果2、遗传机制与非遗传机制(转录重编程)在同一个肿瘤内共存,并呈现出显著的空间异质性。
研究发现:所有8例耐药活检样本均存在转录重编程,包括那些携带多个已知耐药基因突变的样本。
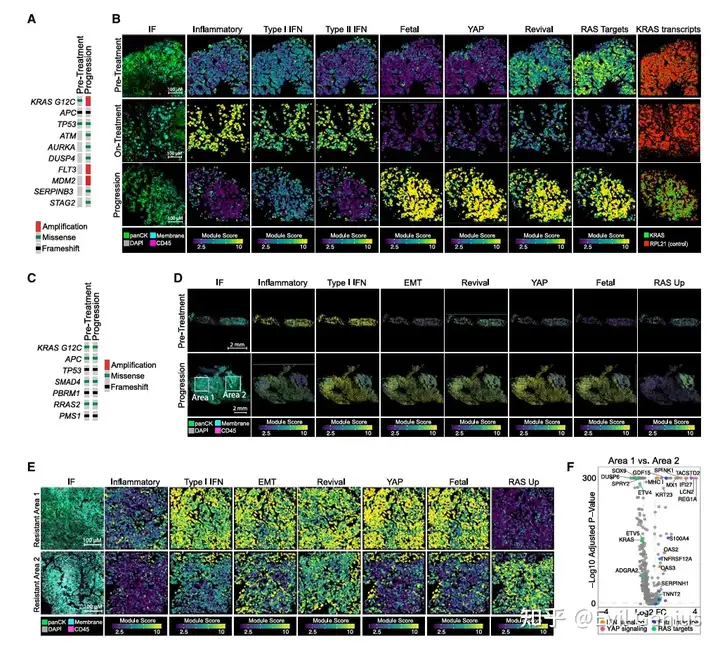
结果3、KRAS抑制后炎症程序的激活是否依赖于肿瘤微环境
KRAS抑制剂治疗后癌细胞出现的转录重编程(尤其是炎症程序)是否需要肿瘤微环境中的其他细胞参与?
微环境中也存在炎症信号,但细胞间直接相互作用有限
在治疗中和耐药样本中,CAF、髓系细胞、淋巴细胞等多个非恶性细胞群体也广泛表现出炎症特征(细胞因子、IFN、JAK-STAT通路)。
但CellChat配体-受体分析显示:免疫细胞与癌细胞之间的直接信号交互较弱(仅在治疗中发现T细胞通过TGFB2-TGFBR2向癌细胞发送信号,而该通路已知与肠道癌细胞耐药相关)。
受限于CosMx平台仅检测950个基因,不能完全排除其他信号通路的参与。
小鼠体内模型:免疫功能不影响耐药进程
利用AKP-G12C类器官(携带Apc、KrasG12C、Trp53突变)移植到免疫正常(C57Bl/6)和免疫缺陷(NSG)小鼠体内。
结果显示:两组小鼠对adagrasib的治疗反应和耐药出现时间无显著差异 → 至少在该模型中,免疫细胞并非耐药所必需。
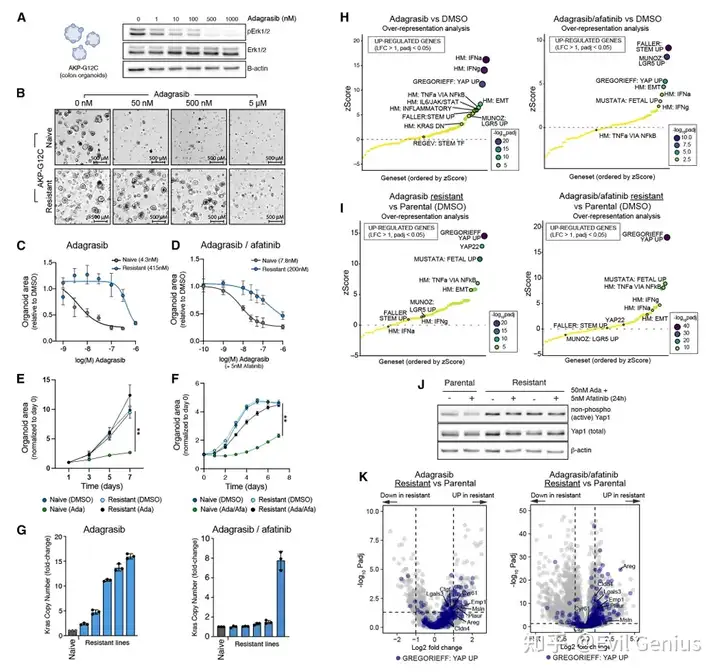
体外类器官模型:完全脱离微环境仍可产生耐药
AKP-G12C类器官在无任何非细胞成分的条件下,单用adagrasib或联合EGFR抑制剂(afatinib)处理:
早期出现生长停滞、ERK磷酸化下降;
持续处理10-12周后出现高度耐药,EC50提高25-100倍;
耐药类器官对sotorasib和RMC-7977(多选择性RAS抑制剂)也产生交叉耐药。
转录组分析显示:
治疗早期(72h):富集肠道干细胞特征 + I型和II型干扰素等炎症程序(与患者活检结果一致);
耐药阶段:炎症程序减弱,YAP和胎儿样特征占主导(无论是否存在Kras扩增)。
耐药类器官中活性YAP(非磷酸化)蛋白水平升高,而急性处理期未见此变化 → 说明YAP激活是长期适应性事件,而非急性反应。
KRAS抑制剂诱导的炎症程序是癌细胞自主性的,可以在完全没有微环境的类器官中被激活。 耐药过程中遗传(如Kras扩增)与非遗传(YAP/胎儿样转录重编程)机制共存,与临床患者表现高度一致。 尽管微环境可能对耐药有调节作用,但癌细胞内在的适应性重编程足以驱动耐药。
结果4、TBK1抑制剂能够阻断KRAS抑制剂治疗后早期的炎症重编程,并与KRAS抑制剂协同抑制肿瘤生长。
已知炎症程序在癌细胞接受靶向或细胞毒性治疗的初始反应阶段被激活。
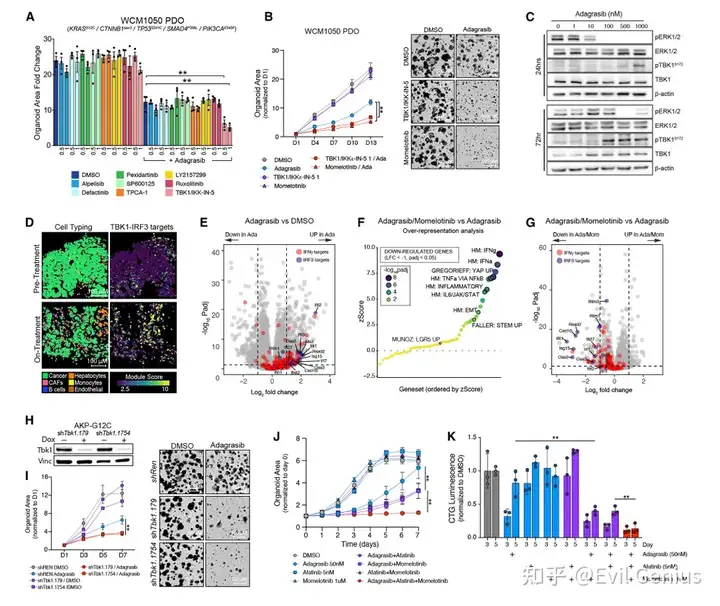
为探究阻断炎症信号是否能抑制早期适应性反应并增强KRAS抑制剂疗效,作者在患者来源的KRAS⁶¹²C突变结直肠癌类器官(PDO) 中进行了小规模药物筛选。
将KRAS抑制剂adagrasib与靶向8种炎症相关激酶(PI3K、FAK、KIT、JNK、IKK、TGFβRI、JAK、TBK1)的小分子抑制剂联合使用。
TBK1是唯一增强疗效的靶点
在筛选的8种抑制剂中,仅TBK1抑制剂(TBK1/IKKε-in-5) 能够延缓adagrasib处理下的类器官生长;TBK1单药无显著效果。
使用FDA批准的双重TBK1/JAK抑制剂momelotinib,同样验证了协同效应。
KRAS抑制剂可激活TBK1-IRF3信号轴
在AKP-G12C类器官中,adagrasib处理后TBK1磷酸化水平呈剂量依赖性升高(24–72小时)。
在KRAS⁶¹³D突变的HCT116细胞中,多选择性RAS抑制剂RMC-7977同样诱导TBK1和IRF3磷酸化。
患者活检样本及类器官转录组分析均显示:KRAS/EGFR抑制后,IRF3靶基因显著上调;而联合momelotinib后,炎症特征(尤其是IRF3靶基因)被显著抑制。
TBK1抑制增强KRAS抑制剂疗效
在多种模型中验证:
小鼠AKP-G12C和AKP-G12D类器官
第二例人KRAS⁶¹²C突变CRC类器官
HCT116细胞
均显示:TBK1抑制剂单药效果微弱,但与KRAS抑制剂联合显著抑制增殖。
基因沉默验证:使用两种独立的诱导型shRNA敲低TBK1,模拟了药物联合的效果 → 证明TBK1抑制本身足以增强adagrasib的活性。
临床转化意义:momelotinib还能够改善当前标准治疗(KRAS/EGFR联合抑制)的疗效。
最后来看看CosMx的分析方法



生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号