Nat. Commun. | 字节Seed团队构建统一有机力场,实现从电子结构到液体性质的跨尺度模拟

Nat. Commun. | 字节Seed团队构建统一有机力场,实现从电子结构到液体性质的跨尺度模拟

DrugOne
发布于 2026-06-02 19:34:03
发布于 2026-06-02 19:34:03
DRUGONE
分子动力学模拟是理解凝聚态体系结构与性质的重要工具,在电池电解液设计、绿色溶剂开发和材料筛选等领域发挥着核心作用。然而,如何仅依赖量子力学计算,在不借助实验参数校正的情况下准确预测液体密度、蒸发焓、电导率等宏观性质,长期以来一直是计算化学领域的重要挑战。
字节Seed团队提出了ByteFF-Pol(ByteDance Polarizable Force Field),一种由图神经网络参数化的可极化有机通用力场。与传统力场和多数机器学习力场不同,ByteFF-Pol完全基于高水平量子化学数据训练,不依赖任何实验数据。研究人员通过引入与ALMO-EDA能量分解分析相匹配的物理驱动势函数、可极化模型以及新的训练策略,实现了从量子力学相互作用到液体热力学和输运性质预测的统一建模。
在近百种纯液体、数百种混合液体以及近5000个电解液体系上的测试结果表明,ByteFF-Pol在密度、蒸发焓、黏度、电导率和扩散系数预测方面均优于现有经典力场和机器学习力场。该工作首次展示了无需实验微调即可从量子力学直接预测复杂液体宏观性质的可行性,为数据驱动材料设计和电解液发现提供了新的技术路线。

分子动力学模拟已经成为现代材料科学和生命科学的重要研究工具。通过追踪原子尺度运动过程,研究人员能够观察实验难以直接测量的微观现象,例如分子扩散、溶剂化结构形成以及相变过程。这些能力使其广泛应用于药物发现、新能源材料设计以及绿色化学等领域。
决定模拟准确性的核心因素是力场。传统力场如AMBER、CHARMM和OPLS通过固定函数形式和经验参数描述原子间相互作用。虽然这些方法计算效率较高,但其准确性很大程度上依赖误差抵消,因此通常需要大量实验数据参与参数优化。
近年来,机器学习力场的发展显著提高了量子化学势能面的拟合能力。然而,纯机器学习方法通常需要海量量子化学数据训练,并且在预测液体等复杂凝聚态体系性质时往往缺乏足够泛化能力。一些性能较好的模型仍然需要引入实验密度等数据进行后期微调。
另一方面,可极化力场能够显式描述电子云对环境变化的响应,被认为是模拟电解液等复杂体系的重要方向。但现有可极化力场仍然存在参数化繁琐、化学空间覆盖有限以及跨体系迁移能力不足等问题。
针对这些挑战,研究人员开发了ByteFF-Pol,希望构建一种兼具量子化学准确性、传统力场效率以及机器学习泛化能力的通用有机力场,实现从微观电子结构到宏观液体性质的直接预测。
方法
ByteFF-Pol采用图神经网络驱动的参数化框架。模型首先从分子图中提取原子和键特征,然后通过边增强图Transformer学习局部化学环境表示,最终生成键参数和非键参数。与传统查表式参数化不同,该方法能够自动适应不同化学环境,提高力场迁移能力。
在非键相互作用方面,ByteFF-Pol将总能量划分为排斥、色散、静电、极化和电荷转移五个组成部分,并使其与ALMO-EDA能量分解结果严格对应。训练阶段,研究人员构建了包含60790个分子二聚体、约600万个构象的大规模量子化学数据集,利用ωB97M-V/def2-TZVPD水平计算及ALMO-EDA分解结果作为监督信号训练模型。
整个训练过程分为预训练、主训练和微调三个阶段。预训练阶段学习化学环境表示;主训练阶段拟合量子化学能量分解项;最后通过扭转势能面微调保证分子内部构象精度。训练完成后,模型可直接输出OpenMM兼容力场参数,并用于大规模分子动力学模拟。
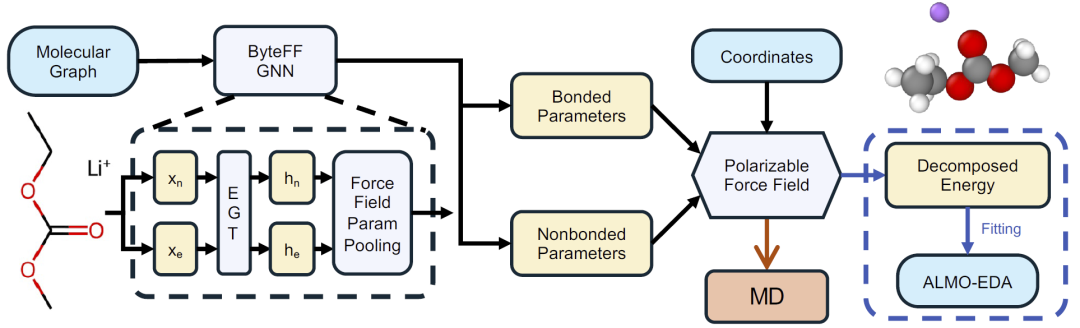
图1:ByteFF-Pol整体框架示意图,包括图神经网络参数生成、ALMO-EDA监督训练以及分子动力学模拟流程。
结果
ByteFF-Pol整体框架实现量子化学驱动参数化
研究人员首先展示了ByteFF-Pol的整体设计框架。系统利用图神经网络直接从分子拓扑生成力场参数,并通过ALMO-EDA分解能量进行监督训练。相比传统依赖人工参数库的方法,该框架能够自动学习不同化学环境下的相互作用规律,实现真正的数据驱动参数化。
模型训练完成后,仅需一次前向传播即可生成整个分子的力场参数,并直接应用于标准分子动力学软件进行模拟。这种设计同时兼顾了参数生成效率和物理可解释性。
准确重建量子力学相互作用能
为了验证微观层面的准确性,研究人员首先评估了ByteFF-Pol对量子化学相互作用能的拟合能力。
在验证集上,模型对总相互作用能预测的平均绝对误差仅为0.36 kcal/mol。进一步分析发现,模型不仅能够准确重现总能量,还能够分别重建色散、静电、极化和电荷转移等不同能量分量。
在中性分子二聚体体系中,ByteFF-Pol能够准确捕获由色散主导的弱相互作用;在锂离子与有机分子体系中,则能够有效描述静电和极化效应。尽管短程极化能存在轻微高估,但不同能量项之间形成合理误差补偿,使总能量仍与DFT结果高度一致。
研究人员进一步将模型推广到多分子簇体系。无论是氢键网络、π-π堆积体系还是离子溶剂化簇,ByteFF-Pol均能准确再现实验和DFT优化结构及结合能,显示出优秀的多体效应泛化能力。
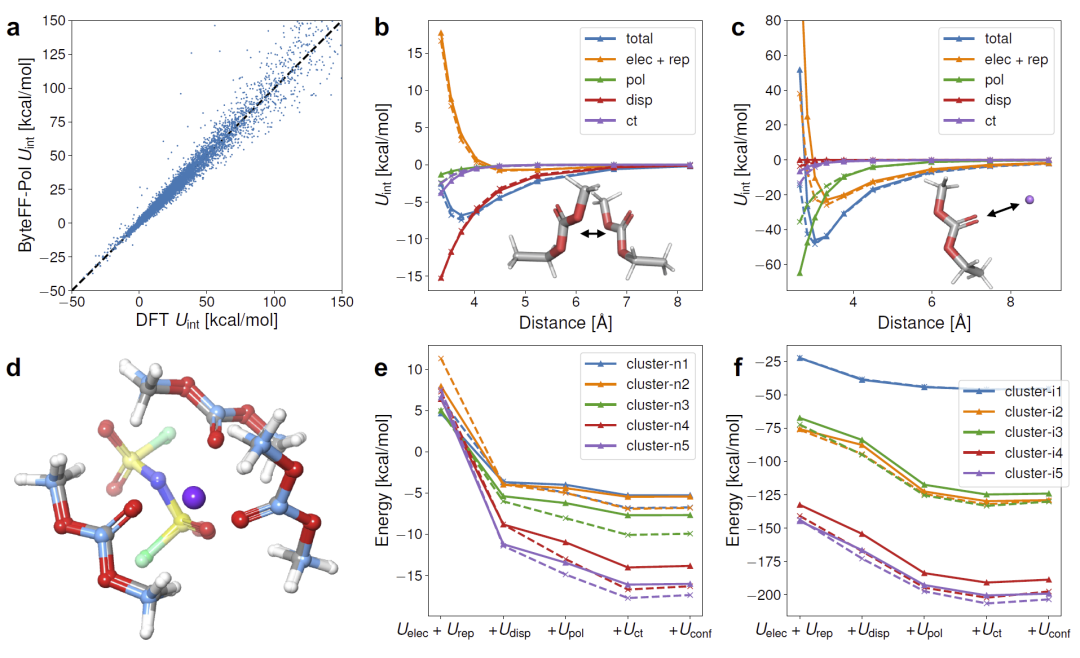
图2:ByteFF-Pol与DFT在二聚体扫描、多分子簇结构及结合能预测中的比较结果。
精准预测液体热力学性质
研究人员随后评估了ByteFF-Pol对宏观液体性质的预测能力。
在96种纯有机液体上,ByteFF-Pol能够准确预测密度和蒸发焓,其表现超过经典力场OPLS-AA和AMOEBA。特别值得注意的是,ByteFF-Pol完全未使用任何实验热力学数据参与训练,而对照力场均包含实验数据参数优化过程。
当进一步引入核量子效应修正后,模型对密度预测的平均误差降低至2.3%,蒸发焓误差降低至7.5%,达到所有比较方法中的最佳水平。
在595个二元液体体系和CRC数据库超过2000种纯液体测试中,ByteFF-Pol仍然保持较高预测精度。研究人员发现,预测性能主要取决于局部原子化学环境是否被训练集覆盖,而非具体分子是否出现在训练集中,这说明模型真正学习到了可迁移的化学规律。
此外,模型还成功预测介电常数、热膨胀系数和等温压缩系数等复杂热力学性质,进一步证明其物理合理性。
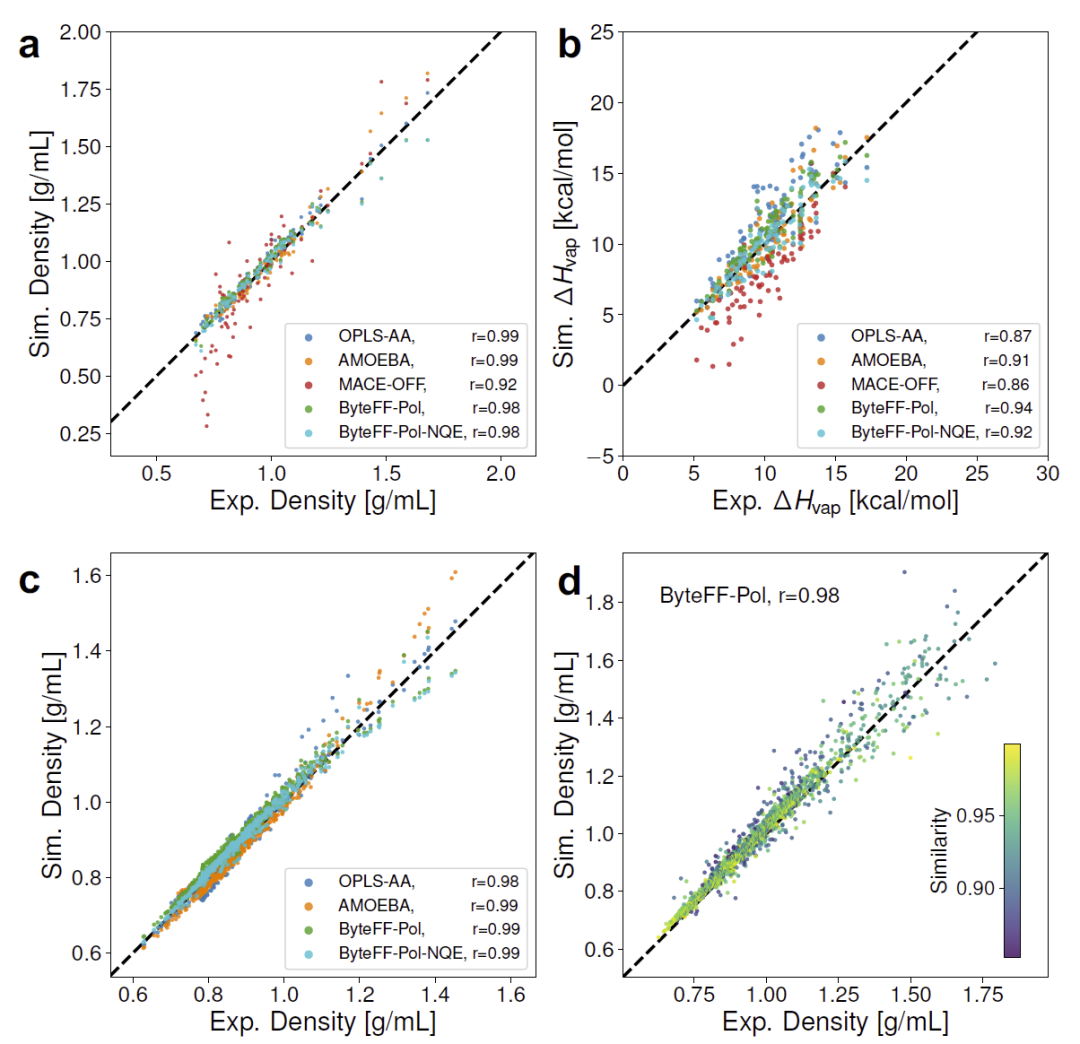
图3:ByteFF-Pol在纯液体、混合液体及大规模CRC数据库中的热力学性质预测结果。
电解液设计中的应用验证
电解液体系是验证力场能力的重要场景,因为其性质同时受到极化、离子相互作用和溶剂结构等多种因素影响。
在88个电解液密度数据集、91个黏度数据集以及4896个电导率数据集上的测试表明,ByteFF-Pol全面优于OPLS-AA、OPLS-AA-SC以及AMOEBA。尤其在电导率预测任务中,模型达到0.95的Pearson相关系数,明显优于所有对照方法。
研究人员进一步展示了实际应用案例。对于不同锂盐浓度体系,ByteFF-Pol不仅能够准确预测电导率数值,还能够正确识别最佳导电浓度对应的位置。对于−40°C至75°C范围内的宽温电解液体系,模型同样保持良好预测能力。
此外,在溶剂扩散系数预测中,ByteFF-Pol能够正确捕获实验趋势,并优于传统固定电荷模型。研究人员认为,这一能力对于未来锂电池电解液设计和筛选具有重要价值。
同时,ByteFF-Pol保持较高计算效率。在A100 GPU上,10000原子体系模拟速度约为40 ns/day,显著快于大多数机器学习力场。
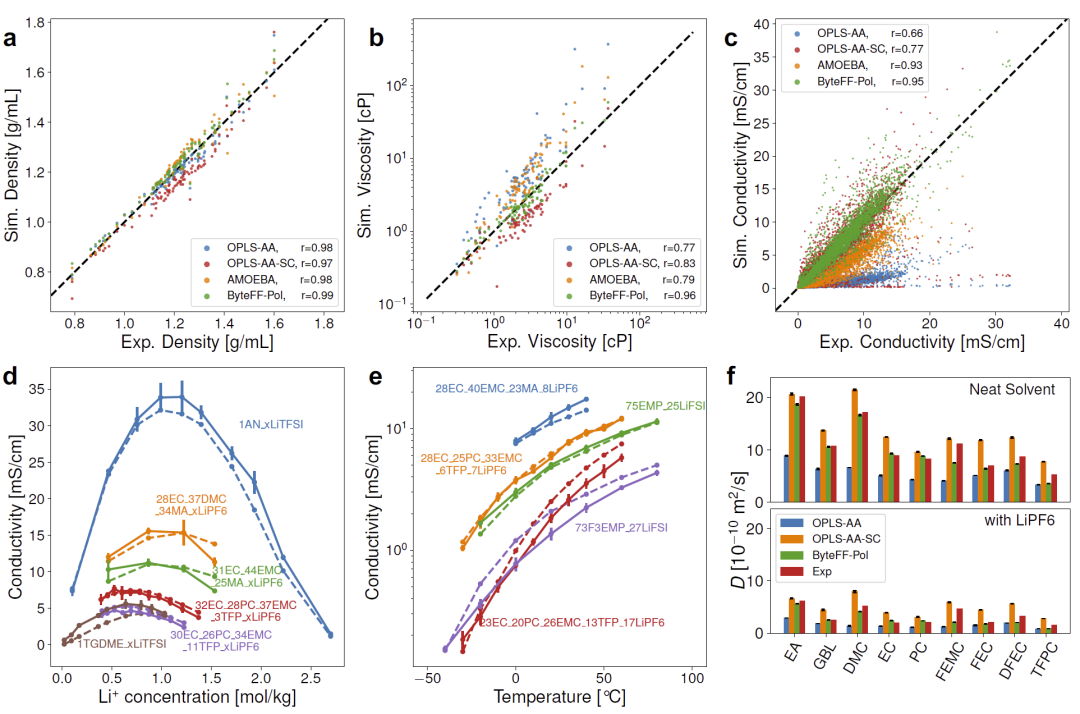
图4:ByteFF-Pol在电解液密度、黏度、电导率、扩散系数及实际电解液设计案例中的应用结果。
讨论
研究人员开发的ByteFF-Pol为长期以来“从量子力学直接预测宏观液体性质”这一目标提供了新的解决方案。与传统依赖实验校正的经验力场不同,ByteFF-Pol完全基于量子化学数据训练,却依然能够准确预测液体密度、蒸发焓、黏度、电导率和扩散系数等关键性质。
该工作的核心创新在于将物理驱动势函数与图神经网络参数化相结合。一方面,通过显式引入极化和电荷转移项,使宏观行为能够自然地从微观相互作用中涌现;另一方面,图神经网络提供了优秀的化学空间泛化能力,使模型能够在未见过的分子体系上实现零样本预测。
与纯机器学习力场相比,ByteFF-Pol无需海量训练数据即可获得优秀迁移性能;与传统力场相比,则摆脱了实验参数依赖。这使其成为真正意义上的“从头算”通用有机力场。
研究人员指出,目前模型对于卤键、σ-hole等各向异性相互作用的描述仍有进一步提升空间。未来通过引入更复杂的势函数形式以及更丰富的量子化学数据,有望进一步提高复杂液体体系模拟精度。
总体而言,ByteFF-Pol成功架起了量子力学与液体宏观性质之间的桥梁,为电解液设计、绿色溶剂开发以及数据驱动材料发现提供了新的基础设施,也展示了字节Seed团队在AI4Science与计算化学领域的重要进展。
整理 | DrugOne团队
参考资料
Zheng, T., Xu, X., Wang, Z. et al. Bridging quantum mechanics to liquid properties via a universal organic force field. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-73566-3
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号