Nat. Comput. Sci. | 生物样本库规模全基因组测序Meta分析统一框架

Nat. Comput. Sci. | 生物样本库规模全基因组测序Meta分析统一框架

DrugAI
发布于 2026-06-04 12:02:34
发布于 2026-06-04 12:02:34
随着英国生物样本库(UK Biobank)、All of Us等大型队列项目陆续完成全基因组测序(WGS),研究人员开始能够系统分析稀有变异在复杂疾病和性状中的作用。然而,由于隐私保护和数据共享限制,不同生物样本库之间往往难以直接共享个体水平数据,从而限制了联合分析的统计效能。传统Meta分析工具虽然能够整合多个队列结果,但在处理生物样本库规模的WGS数据时面临存储开销大、计算效率低以及难以整合功能注释信息等问题。
研究人员开发了MetaSTAARlite,这是一个面向生物样本库规模全基因组和全外显子组测序Meta分析的一体化工具。MetaSTAARlite能够自动生成资源高效的汇总统计量,利用稀疏矩阵实现高效计算,并支持整合多种功能注释开展功能增强型稀有变异关联分析。通过对UK Biobank和All of Us数据的验证,研究人员发现MetaSTAARlite在计算时间、内存占用和存储需求方面均随样本量线性增长,同时能够获得与个体水平数据合并分析几乎一致的结果。研究结果表明,该工具不仅显著降低了超大规模基因组Meta分析的计算成本,而且能够保持与联合分析相当的统计效能,为未来跨生物样本库稀有变异研究提供了高效解决方案。

近年来,大规模全基因组测序项目极大推动了人类遗传学的发展。相比常见变异,稀有变异通常具有更大的效应值,因此对于理解复杂疾病遗传机制和发现潜在治疗靶点具有重要价值。然而,由于单个研究队列中的稀有变异携带者数量有限,研究人员往往需要联合多个大型队列进行分析。
虽然个体水平数据合并分析能够获得最高统计效能,但现实中受到隐私保护法规、数据访问限制以及跨机构协作成本等因素影响,不同研究队列之间通常无法共享原始基因型数据。因此,基于汇总统计量的Meta分析成为更加现实的选择。
此前研究团队开发的STAARpipeline已经能够在单个生物样本库内高效开展稀有变异分析,并利用功能注释提高检测能力。然而,该框架仍依赖个体水平数据。随后出现的MetaSTAAR实现了稀有变异Meta分析,但随着样本量增长,其存储和计算负担迅速增加。研究人员因此提出MetaSTAARlite,希望在保持分析精度的同时,进一步提高可扩展性和资源利用效率。
方法
MetaSTAARlite整体由三个核心步骤组成。首先,在每个参与研究中建立广义线性混合模型,校正群体结构和样本亲缘关系,并生成变异水平汇总统计量以及稀疏连锁不平衡矩阵(sparse LD matrix)。其次,将多个研究队列产生的汇总统计量进行整合,通过精确重建得分统计量的协方差矩阵,实现与个体水平数据联合分析等价的统计推断。最后,利用MetaSTAAR-O开展功能增强型稀有变异Meta分析,并结合多个定量功能注释作为权重提高检测能力。
为了提高效率,MetaSTAARlite在整个流程中采用稀疏矩阵表示,包括稀疏遗传关系矩阵(GRM)和稀疏基因型矩阵,从而显著降低内存消耗和计算复杂度。同时,平台支持蛋白编码区、非编码调控区以及用户自定义分析单元,并自动生成可视化结果和后续分析报告。
结果
MetaSTAARlite构建统一的生物样本库级Meta分析框架
研究人员首先介绍了MetaSTAARlite的整体工作流程。系统从每个研究队列的基因型、表型和协变量数据出发,进行功能注释、祖源主成分分析和稀疏GRM构建。随后自动生成变异层面的汇总统计量和稀疏LD矩阵,并利用MetaSTAAR-O进行功能增强型Meta分析。分析完成后,系统还能自动识别驱动关联信号的功能注释以及潜在致病变异,并生成结果可视化和条件分析报告。
与传统流程相比,MetaSTAARlite将数据预处理、统计分析和结果解释整合到统一框架中,极大降低了大规模遗传学Meta分析的技术门槛。
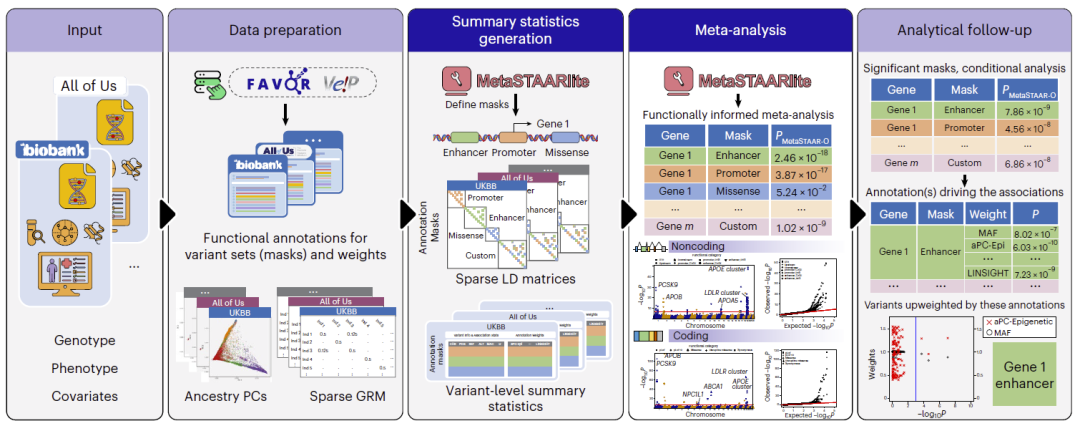
图1:MetaSTAARlite整体框架,包括输入数据准备、功能注释、汇总统计量生成、Meta分析以及结果追踪与解释流程。
计算效率和扩展性显著提升
研究人员利用UK Biobank外显子组测序数据对MetaSTAARlite进行基准测试,并与MetaSTAAR和Raremetal2进行比较。
结果显示,在30万样本规模下,MetaSTAARlite的峰值内存消耗相比MetaSTAAR降低332倍,相比Raremetal2降低1386倍;运行时间分别缩短24倍和2206倍。即使在44.6万个样本、22994个变异的最大测试规模下,MetaSTAARlite仍能在不到49秒内完成汇总统计量生成,而峰值内存不足1 GB。相比之下,Raremetal2即使分配768 GB内存仍无法完成任务。
进一步分析发现,MetaSTAARlite的运行时间和内存占用均随样本量近似线性增长,显示出优异的扩展能力。
结显著降低存储需求
为了评估存储效率,研究人员将190110名UK Biobank参与者随机划分为三个研究队列,并开展全基因组Meta分析。
结果表明,即使同时整合12种功能注释,编码区分析仅需约2.40 GB存储空间,非编码区分析约需13.32 GB。更重要的是,稀疏LD矩阵仅占总存储需求的7.7%至17.8%。随着样本量增加,存储需求依然呈线性增长。
研究人员认为,这种设计使得LD矩阵不再成为稀有变异Meta分析的主要瓶颈,为未来百万样本规模分析奠定了基础。
Meta分析结果与联合分析高度一致
研究人员以总胆固醇(TC)为例,对三个随机划分的数据集开展Meta分析,并与基于全部190110名参与者的STAARpipeline联合分析结果进行比较。
结果显示,无论是编码区还是非编码区分析,MetaSTAARlite获得的P值分布均保持良好校准。在编码区分析中,共发现58个全基因组显著关联;在非编码区分析中发现88个显著关联位点。更重要的是,MetaSTAARlite与联合分析得到的结果几乎完全重合。显著位点的log10(P)值相关系数超过0.999。
此外,MetaSTAAR-O相比传统Burden、SKAT和ACAT-V方法通常获得更小的P值,并发现了一些其他方法未能检测到的关联信号。
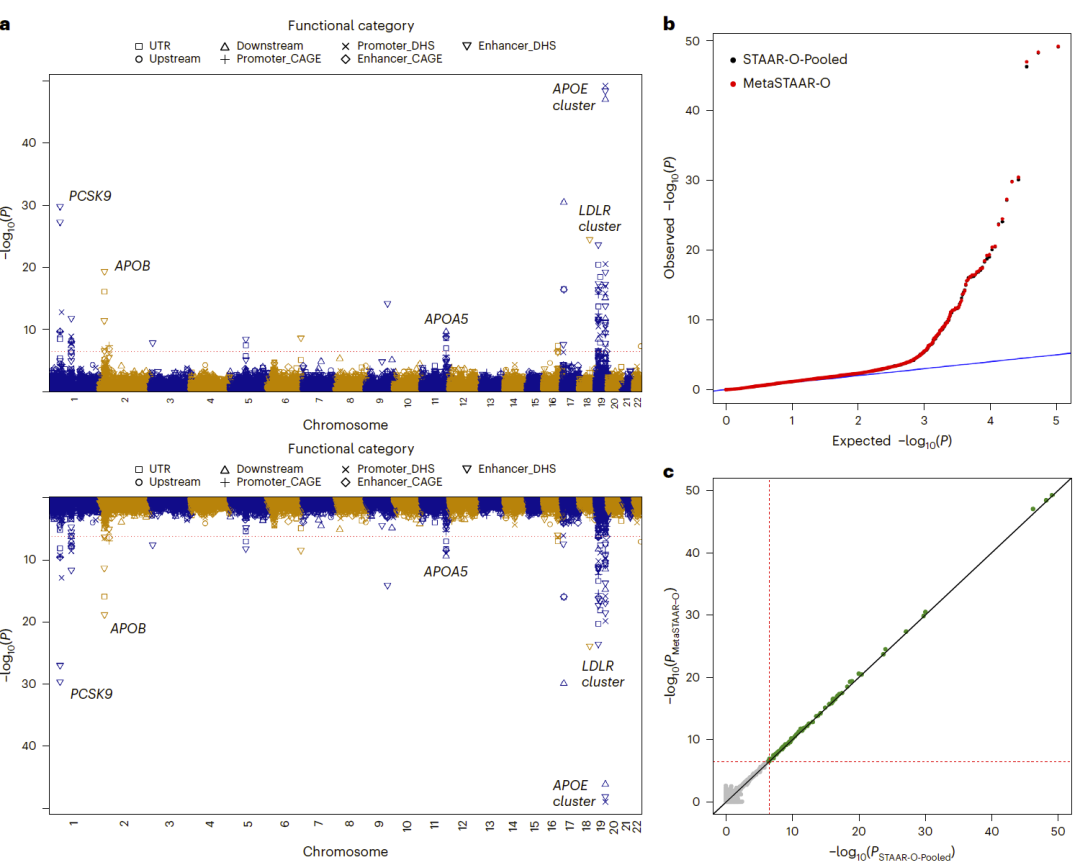
图2:MetaSTAARlite与STAARpipeline联合分析结果比较,包括Miami图、QQ图以及显著位点P值相关性分析。
跨生物样本库Meta分析验证
研究人员进一步整合UK Biobank和All of Us两个大型队列,对总胆固醇、身高、估算肾小球滤过率(eGFR)、血钙以及高LDL胆固醇等五个性状开展分析,总样本量达到692445人。
在全基因组范围内,MetaSTAARlite分别发现165、536、117、38和94个显著关联基因。整个分析过程中平均仅需95.4 CPU小时,峰值内存始终低于1 GB。对于每个性状,全基因组汇总统计量仅需约1–2 GB存储空间。
研究结果证明,MetaSTAARlite能够在多祖源、大规模生物样本库环境下稳定运行,并保持优秀的统计效能和资源利用效率。
讨论
研究人员开发的MetaSTAARlite为生物样本库时代的稀有变异Meta分析提供了一套完整解决方案。与传统依赖个体水平数据的联合分析相比,该方法能够在严格保护参与者隐私的前提下,实现几乎完全一致的统计结果。
该工具最大的创新在于充分利用稀疏矩阵表示。通过稀疏GRM、稀疏基因型矩阵以及稀疏LD矩阵的统一设计,MetaSTAARlite大幅降低了内存、存储和计算开销,使百万样本规模分析成为现实。与此同时,功能注释加权机制进一步提高了稀有变异检测能力,有助于发现潜在因果变异和调控元件。
研究人员还强调,MetaSTAARlite不仅支持编码区和非编码区分析,还支持用户自定义分析单元以及条件Meta分析,并能够自动生成Manhattan图、QQ图和结果汇总文件。因此,该平台兼具灵活性和易用性。
未来,MetaSTAARlite有望进一步扩展到生存分析、纵向数据分析、多性状联合分析以及自适应变异集合分析等更复杂场景。随着全球生物样本库持续扩张,该工具有望成为跨队列全基因组测序Meta分析的重要基础设施,为复杂疾病遗传学研究和精准医学发展提供关键支持。
整理 | DrugOne团队
参考资料
Kumarasinghe, Y., Williams, J., Yuan, Y. et al. MetaSTAARlite: an all-in-one tool for biobank-scale whole-genome sequencing meta-analysis. Nat Comput Sci (2026).
https://doi.org/10.1038/s43588-026-00995-x

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号