文献分享--CD4+ T细胞通过IL-3和TNF依赖性血管损伤抑制肿瘤生长
原创
文献分享--CD4+ T细胞通过IL-3和TNF依赖性血管损伤抑制肿瘤生长
原创
追风少年i
修改于 2026-06-23 09:00:03
修改于 2026-06-23 09:00:03
作者,Evil Genius
生物信息分析需要补充很多的基础知识和生物学背景,生信分析,包括生物和信息两部分,计算机语言可以帮助我们分析出结果,而生物学背景帮助我们判断结果是否准确,赋予结果生物学意义,两者不可分割,光有信息,类似公司标准化跑流程,无法结合生物学背景赋予其生物学意义,光有生物学也不行,因为我们需要工具对数据进行处理。
举一个简单的例子,空间注释,通过计算机分析可以得到巨噬细胞和肿瘤细胞在某个部位相互临近,而两者邻近的生物学意义,是促进肿瘤扩展还是抑制肿瘤进展,就需要我们的生物学背景来判断。
今日参考文献

知识积累
癌细胞通常会产生与正常细胞不同的分子(抗原),这些分子可被适应性免疫系统识别。大多数癌症免疫治疗策略,包括免疫检查点阻断和细胞疗法,都利用淋巴细胞直接杀伤表达特定抗原靶标并将其“呈递”在表面的肿瘤细胞的能力。然而,癌症会通过限制淋巴细胞进入肿瘤组织,或通过最初罕见的、不表达或不呈递该抗原的变异细胞选择性存活来逃避免疫清除。
现有疗法的困境:免疫治疗对突变少、缺少新抗原的肿瘤效果差,且肿瘤微环境本身具有“防御力”,导致治疗抵抗。
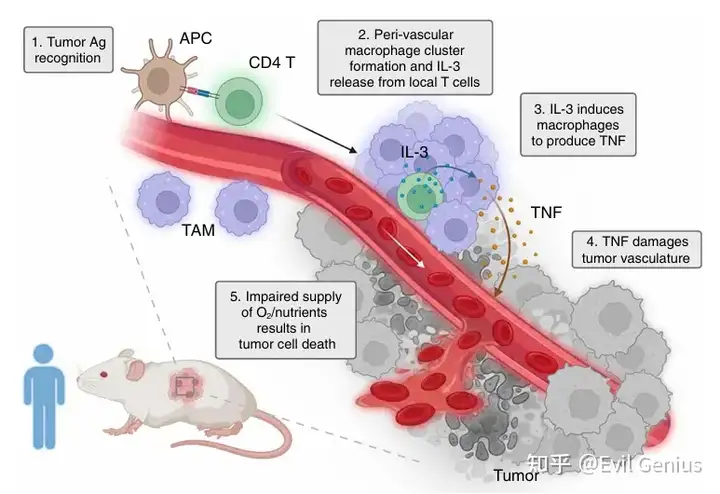
CD4⁺ T细胞的角色:CD4⁺ T细胞在免疫中作用复杂(既可促癌也可抑癌)。它们不直接识别肿瘤细胞,而是识别由其他抗原呈递细胞呈递的肿瘤抗原。
CD4⁺ T细胞的双面性:它既能抗肿瘤,也能促肿瘤,在癌症中的作用十分复杂。
本研究的切入点:作者专门寻找CD4⁺ T细胞中纯粹抑制肿瘤的机制。
核心发现:发现了一条全新的通路——CD4⁺ T细胞不直接攻击癌细胞,而是通过分泌IL-3“教育”巨噬细胞产生TNF,转而破坏肿瘤内部的血管,切断供血,最终“饿死”或杀死肿瘤细胞。
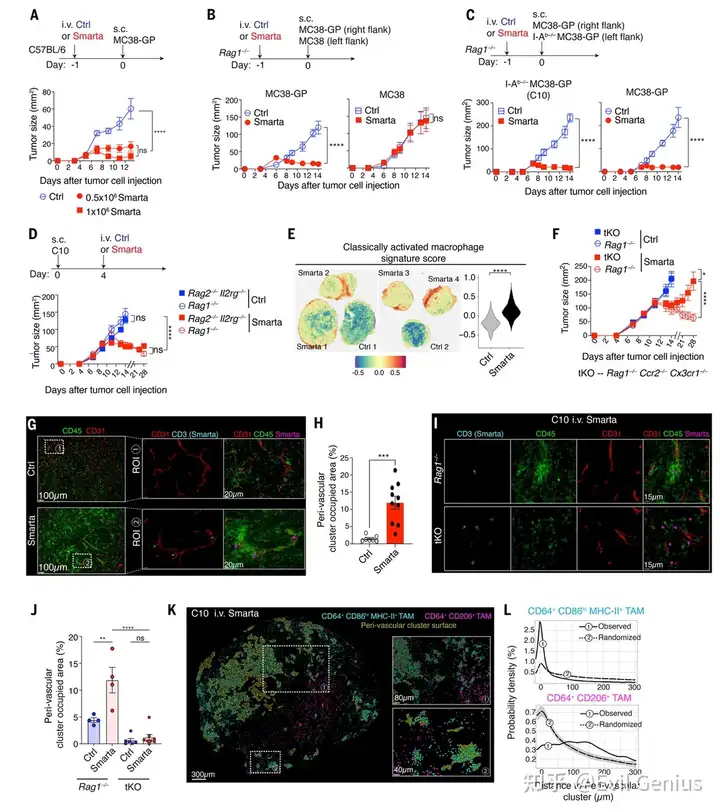
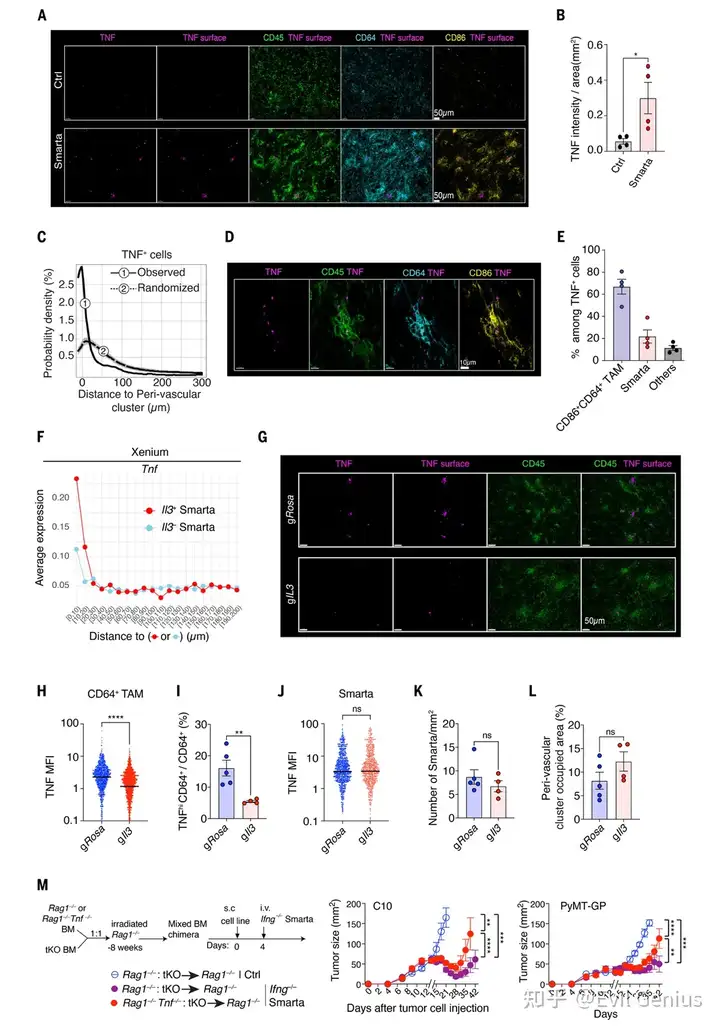
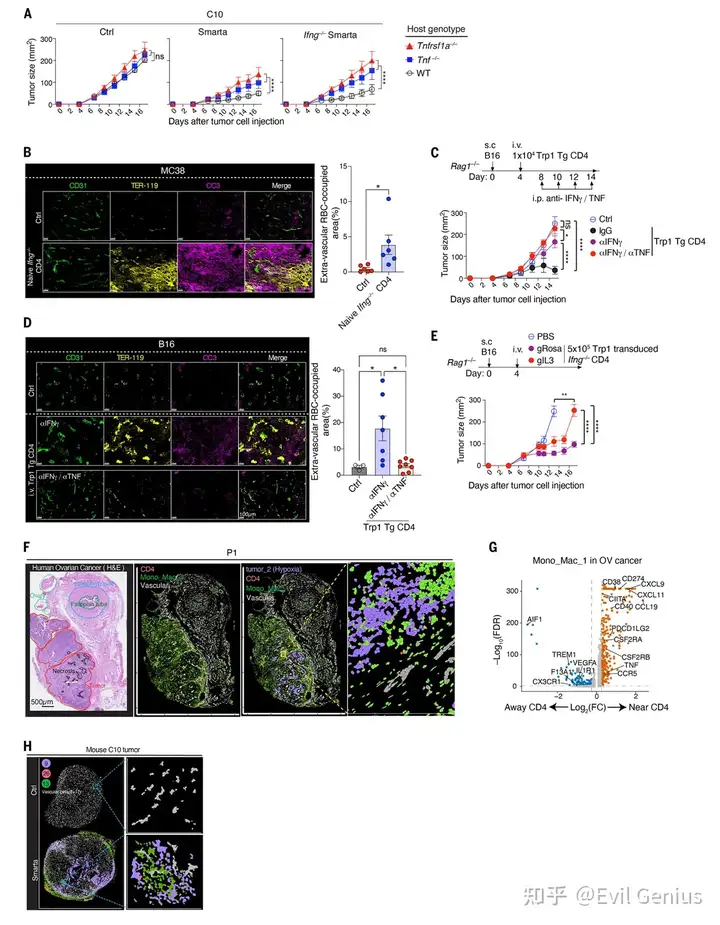
结果1、CD4⁺ T细胞介导的肿瘤控制独立于其他淋巴细胞和直接细胞毒性
CD4⁺ T细胞可独立抗肿瘤:Smarta CD4⁺ T细胞能特异性抑制表达抗原(GP)的肿瘤,该作用不依赖CD8⁺ T、B、NK等其他淋巴细胞,也不要求肿瘤细胞自身表达MHC II(即不需要T细胞直接识别肿瘤细胞表面的抗原)。
抗肿瘤作用依赖髓系细胞:通过阻断单核细胞向肿瘤的募集(tKO小鼠),CD4⁺ T细胞的抗肿瘤效果基本消失,证明其依赖单核-巨噬细胞谱系细胞。
CD4⁺ T细胞在血管周围“集结”巨噬细胞:CD4⁺ T细胞进入肿瘤后,会在血管周围形成由大量CD64⁺巨噬细胞组成的“血管周围簇”。这种结构的形成依赖局部抗原和单核细胞募集。
巨噬细胞出现空间上的功能分化:在血管周围簇内的巨噬细胞高表达炎症型标志物(CD86、MHC-II),而表达调节型标志物(CD206)的巨噬细胞则分布在这些簇之外。这解释了转录组中混合信号的来源。
CD4⁺ T细胞不直接杀癌细胞,而是通过招募并“教育”巨噬细胞在肿瘤血管周围聚集,为后续通过TNF损伤血管、抑制肿瘤奠定基础。
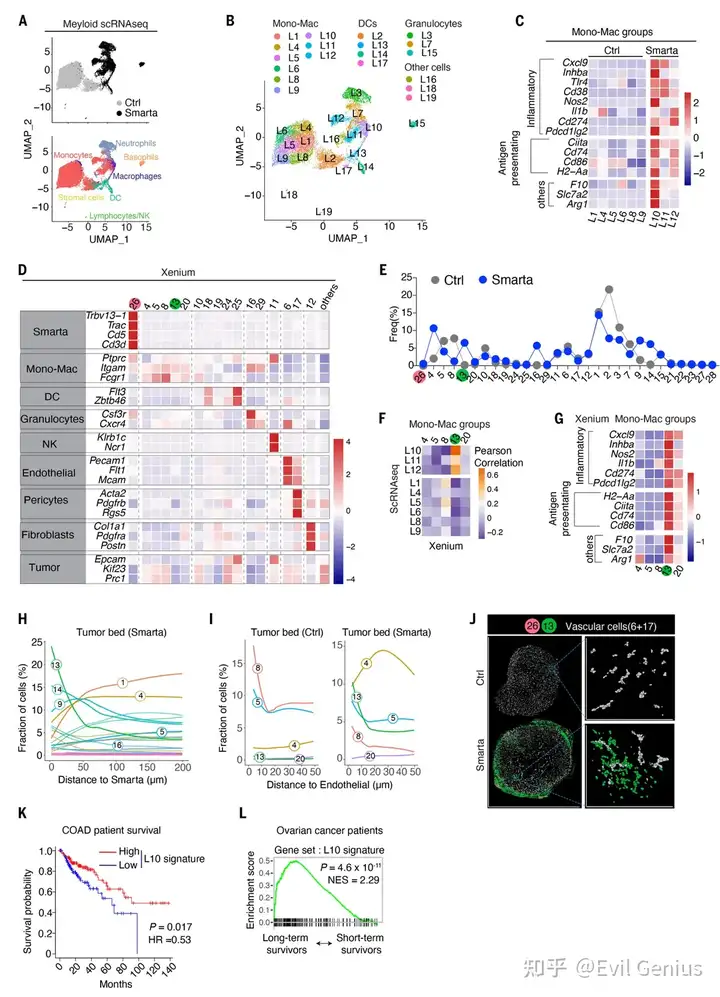
结果2、CD4⁺ T细胞诱导髓系细胞的组织转录组变化
CD4⁺ T细胞诱导特定巨噬细胞亚群(L10/Group 13):通过单细胞RNA测序(scRNA-seq) 发现,Smarta CD4⁺ T细胞的存在使肿瘤内出现一群独特的单核-巨噬细胞(L10群组),高表达炎症性基因(如Cxcl9、Nos2)和MHC II抗原呈递基因,同时具有混合特征(部分细胞共表达Nos2和Arg1)。
空间定位验证:通过10x Genomics Xenium原位转录组学技术(单细胞/高分辨率空间转录组)结合IBEX多重成像技术,确认这群炎症性巨噬细胞(Group 13)在空间上与Smarta T细胞和血管内皮细胞距离最近,即它们正是之前在IBEX成像中看到的“血管周围簇”中的巨噬细胞。
临床相关性:该炎症性巨噬细胞基因特征(L10 signature)在TCGA人类癌症转录组数据库的结肠腺癌分析中与更长的患者生存期相关,并且在接受CTLA-4和PD-1/PD-L1联合免疫治疗的卵巢癌患者中,长期存活者体内富集这一特征。
CD4⁺ T细胞在肿瘤内诱导出一群特殊的炎症性巨噬细胞(血管周围簇),这群细胞的基因特征与人类癌症患者更好的预后和免疫治疗应答相关,进一步证实了小鼠模型中发现的机制具有临床转化潜力。
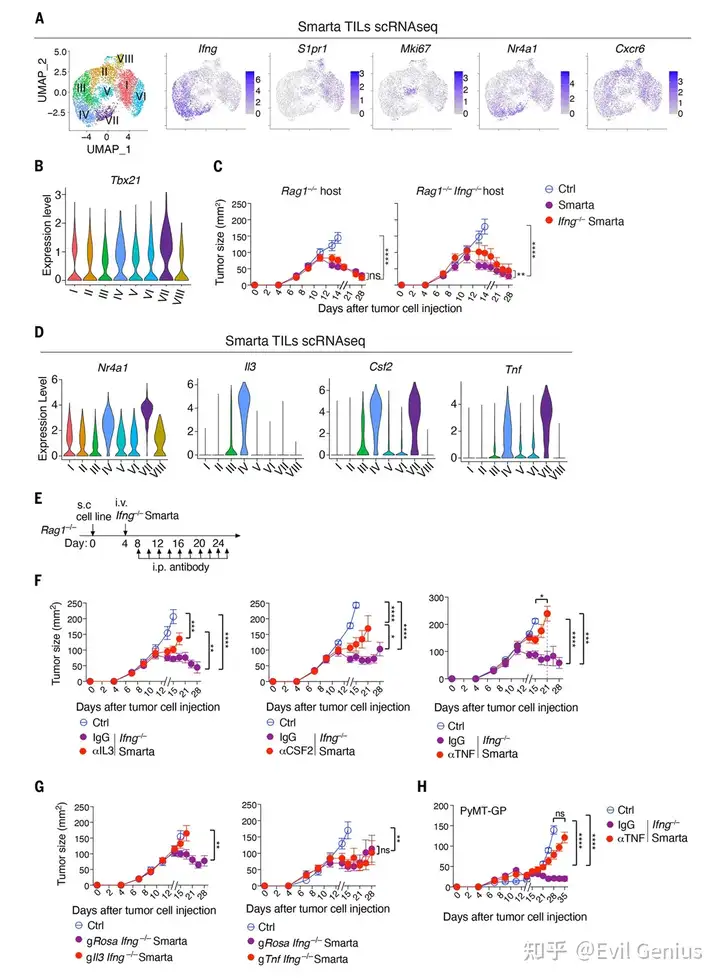
结果3、Smarta细胞的特征分析
Smarta CD4⁺ T细胞呈现Th1样特征:单细胞测序显示,肿瘤内的Smarta T细胞表达Ifng、Tbx21和Nr4a1,证明它们被肿瘤抗原激活并呈现保守的Th1样反应。
肿瘤控制不完全依赖IFN-γ:即使IFN-γ完全缺失,Smarta细胞仍能控制肿瘤生长,说明存在IFN-γ非依赖性的抗肿瘤通路(既往研究多关注IFN-γ通路)。
关键细胞因子鉴定——IL-3(T细胞来源)和TNF(宿主细胞来源):
抗体阻断实验证明IL-3、CSF2和TNF均不可或缺。
CRISPR基因敲除证明:Smarta T细胞自身产生的IL-3是必需的,但T细胞自身产生的TNF不是必需的。
在TNF缺陷宿主中,肿瘤控制受损,证明效应TNF由宿主细胞(即巨噬细胞)产生。
TNF是长期肿瘤控制的关键:即使IFN-γ通路完整,长期肿瘤控制仍依赖TNF;IFN-γ仅能提供短暂的抑制效应。
CD4⁺ T细胞通过分泌IL-3,激活宿主巨噬细胞产生TNF,TNF进而发挥抗肿瘤效应——而T细胞自身产生的IFN-γ和TNF均非必需,阐明了一条由IL-3 → 巨噬细胞TNF介导的全新CD4⁺ T细胞抗肿瘤通路。
TNF的主要来源是巨噬细胞:在CD4⁺ T细胞形成的血管周围结构中,TNF主要由CD64⁺ CD86⁺炎症性巨噬细胞产生,T细胞自身也表达少量TNF。
IL-3敲除实验证明因果关系:用CRISPR敲除Smarta T细胞的Il3基因后,血管周围结构依然形成,但巨噬细胞的TNF表达显著降低,肿瘤控制也随之消失。这证明:T细胞产生的IL-3是驱动巨噬细胞产生TNF的“命令信号”。
骨髓嵌合体实验进一步锁定巨噬细胞来源的TNF:通过构建混合骨髓嵌合体,使浸润肿瘤的单核-巨噬细胞特异性缺乏TNF能力,结果肿瘤控制受损。这直接证明效应TNF确实来自巨噬细胞(而非其他宿主细胞)。
完整信号通路
CD4⁺ T细胞 → 分泌IL-3 → 血管周围巨噬细胞 → 产生TNF → 损伤肿瘤血管 → 肿瘤细胞死亡
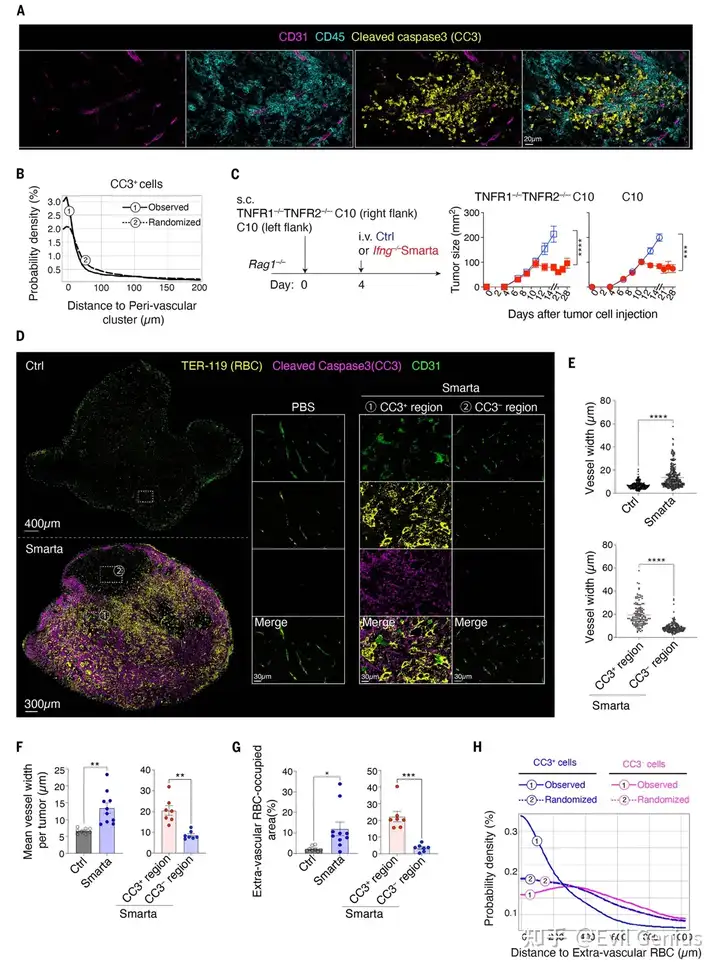
结果4、CD4⁺ T细胞诱导TNF依赖性的肿瘤血管破坏
TNF不直接作用于肿瘤细胞:用CRISPR敲除肿瘤细胞的TNFR1和TNFR2受体后,CD4⁺ T细胞的抗肿瘤效果完全不受影响,证明TNF并非直接杀伤癌细胞,而是通过作用于基质细胞间接起效。
TNF靶向肿瘤血管系统:在CD4⁺ T细胞处理组中,肿瘤内血管出现明显的扩张,并且红细胞从血管内渗漏到周围肿瘤组织中,表明血管完整性被破坏。
血管损伤导致肿瘤细胞死亡:
在血管周围的凋亡(CC3⁺)区域,血管扩张和红细胞渗漏显著;
而在非凋亡(CC3⁻)区域或对照组中,这些现象几乎不存在。
用抗TNF抗体阻断TNF后,血管损伤和细胞凋亡均被阻止。
该机制具有普适性:在PyMT-GP乳腺癌肿瘤模型中也观察到了完全相同的血管扩张、红细胞渗漏和肿瘤抑制现象,证明这不是某一特定肿瘤类型的特有效应。
完整信号通路总结:
肿瘤特异性CD4⁺ T细胞 → 分泌IL-3 → 募集并编程血管周围巨噬细胞 → 巨噬细胞产生TNF → TNF攻击肿瘤血管(内皮细胞损伤、血管通透性增加) → 血管破裂、红细胞渗漏、组织缺氧 → 肿瘤细胞大量死亡 → 肿瘤消退
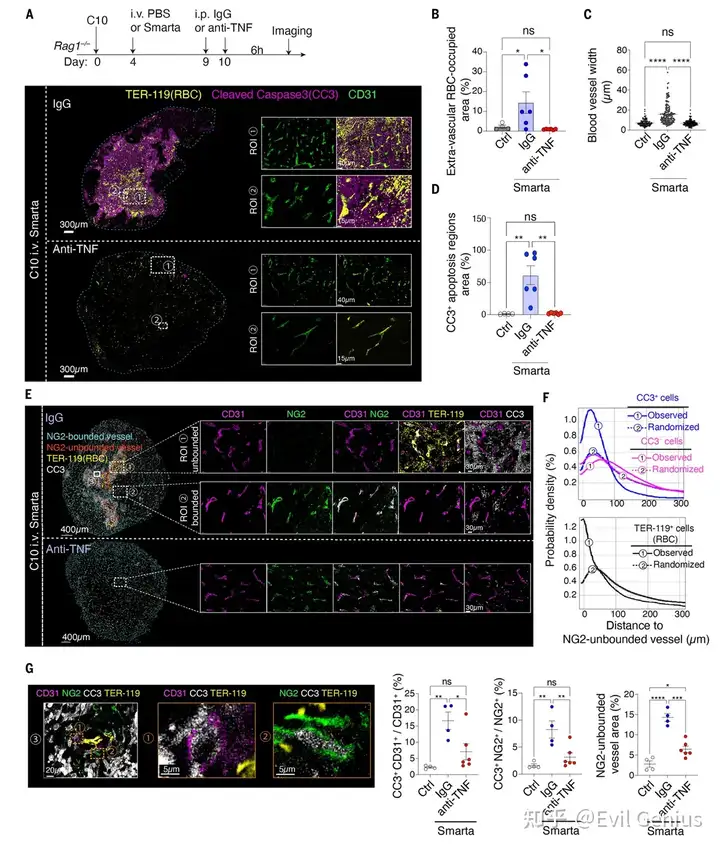
血管损伤的细胞层面,揭示了TNF攻击肿瘤血管的具体靶细胞和病理过程
TNF的靶细胞是血管内皮细胞和周细胞:Xenium分析显示,肿瘤血管的内皮细胞和周细胞均高表达TNF受体(Tnfrsf1a),是TNF的直接作用靶点。
TNF导致血管结构崩解:
在正常(非凋亡)区域,周细胞紧密包绕血管(“bounding”),维持血管结构完整;
在凋亡(CC3⁺)区域,血管显著扩张,周细胞与内皮细胞之间的紧密联系丧失(周细胞脱离、内皮细胞裸露),血管壁完整性被破坏。
血管细胞自身发生凋亡:CD31⁺内皮细胞和NG2⁺周细胞中CC3⁺凋亡信号增加,证明TNF直接诱导了这些血管支持细胞的死亡。
红细胞外渗:血管结构破坏后,红细胞从血管内漏出到周围肿瘤组织中,表明血管屏障功能彻底丧失。
抗TNF抗体可部分逆转上述损伤:验证了所有病理变化均依赖TNF信号。
机制深化
CD4⁺ T细胞 → IL-3 → 巨噬细胞 TNF → 结合内皮细胞/周细胞上的TNFR1 → 诱导血管支持细胞凋亡 → 周细胞脱离、血管壁破裂 → 红细胞渗漏、组织缺氧 → 肿瘤细胞大量死亡
血流灌注破坏:番茄凝集素灌注实验显示,Smarta处理组肿瘤的血管灌注严重受损,且损伤部位与血管周围髓系簇的位置高度一致。
肿瘤细胞缺氧:在红细胞渗漏区域周围出现大范围HIF1A(缺氧标志物)阳性区域,证明血管损伤导致肿瘤组织严重缺氧。
缺氧无法阻止肿瘤细胞死亡:在肿瘤细胞中,HIF1A信号与凋亡标志物CC3完全重叠,说明虽然肿瘤细胞启动了缺氧适应反应,但损伤过于严重,仍无法避免大量死亡。
受体-配体遗传学验证:
将TNF受体充足的骨髓移植到TNF受体缺陷(Tnfrsf1a–/–)的宿主中,CD4⁺ T细胞的抗肿瘤效果消失。
由于供体骨髓细胞(包括巨噬细胞)对TNF有反应,但宿主血管细胞缺乏TNF受体,因此唯一合理的解释就是TNF直接作用于宿主来源的血管细胞(而非巨噬细胞自身或其他造血细胞)。
血管细胞的独特性:血管细胞是肿瘤床内唯一广泛分布的非造血细胞(成纤维细胞仅位于肿瘤周边),这与TNF作用的解剖学位置完全吻合。
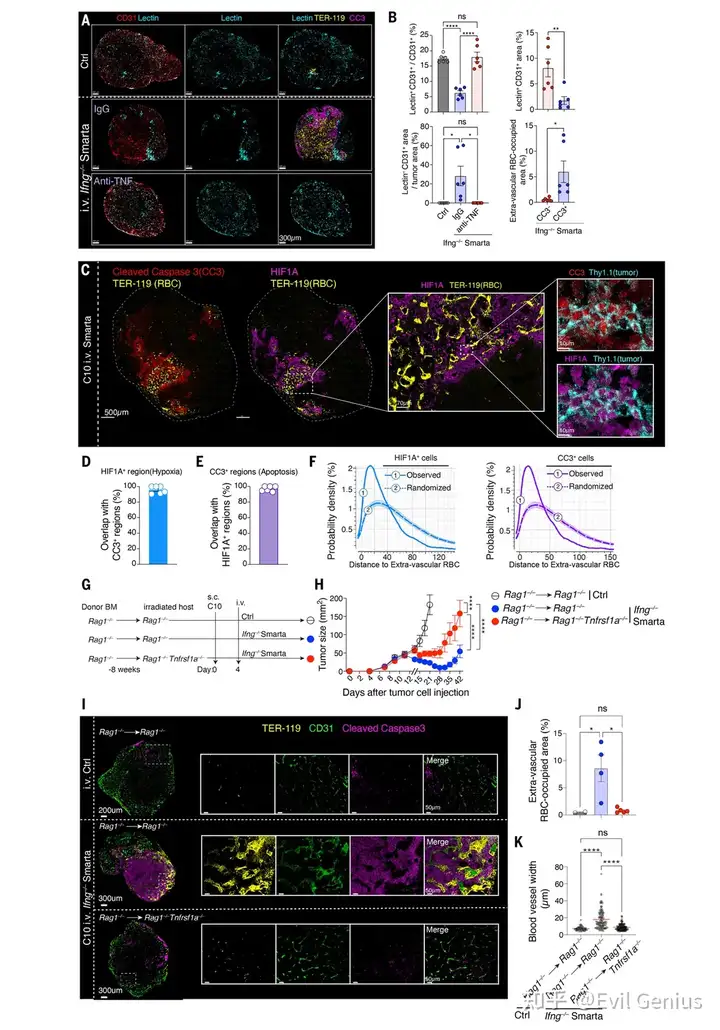
完整机制链
肿瘤特异性CD4⁺ T细胞 → 分泌IL-3 → 募集并编程血管周围巨噬细胞 → 巨噬细胞产生TNF → TNF结合血管内皮细胞/周细胞上的TNFR1 → 周细胞脱离+内皮细胞凋亡 → 血管破裂、灌注中断 → 肿瘤缺氧 → 大规模肿瘤细胞死亡 → 肿瘤消退
机制在免疫健全宿主中同样有效:在免疫系统完整的小鼠中,该通路同样能控制肿瘤,证明其并非免疫缺陷环境下的“人为现象”。
不限于特定TCR或特定抗原:
多克隆CD4⁺ T细胞(非转基因)也能识别肿瘤内源性抗原并触发同样的血管损伤效应。
在B16黑色素瘤+Trp1抗原系统中,同样验证了该机制。
IL-3-TNF通路的普适性:在B16模型中,CRISPR敲低Il3同样破坏了CD4⁺ T细胞的抗肿瘤能力,证明IL-3→TNF通路在不同肿瘤模型中通用。
人类肿瘤中的相关性(最关键):
在结直肠癌和卵巢癌患者样本中,同样发现了CD4⁺ T细胞 + L10样巨噬细胞 + 血管的空间共定位模式。
人类肿瘤中,邻近CD4⁺ T细胞的巨噬细胞高表达炎症和抗原呈递基因(与小鼠L10特征一致)。
缺氧样肿瘤细胞与CD4⁺ T细胞/巨噬细胞聚集体空间重叠,且与H&E染色的坏死区域一致——完整复现了小鼠中的“血管周围簇→血管损伤→缺氧→肿瘤坏死”链条。
抗原激活的CD4⁺ T细胞 → 分泌IL-3 → 诱导血管周围巨噬细胞产生TNF → 破坏肿瘤血管(周细胞脱离+内皮细胞死亡)→ 血流中断、缺氧 → 肿瘤细胞大量死亡 → 肿瘤生长受控。该机制在多种小鼠肿瘤模型中普遍存在,且在人类结直肠癌和卵巢癌中具有相似的空间组织学证据,具有重要的临床转化潜力。
最后来看看方法


生活很好,有你更好。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
原创声明:本文系作者授权腾讯云开发者社区发表,未经许可,不得转载。
如有侵权,请联系 cloudcommunity@tencent.com 删除。
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号