上海药物所团队打造PBCNet2.0 :让 AI 从原子层面读懂蛋白–配体结合

上海药物所团队打造PBCNet2.0 :让 AI 从原子层面读懂蛋白–配体结合

DrugIntel
发布于 2026-06-24 14:05:37
发布于 2026-06-24 14:05:37

PBCNet2.0 深度解读:笛卡尔张量等变网络如何把"FEP 级"亲和力预测带入高通量时代
期刊 Nature Chemical Biology(2026) · DOI 10.1038/s41589-026-02241-x 团队 中科院上海药物所(SIMM)郑明月 / 张素林 / 王明亮 等 代码github.com/YuJie-0202/PBCNet2.0
摘要
结合亲和力预测是分子探针开发与先导化合物优化的核心瓶颈:物理方法(以 Schrödinger FEP+ 为代表)精度可靠但算力昂贵、设置复杂;深度学习高通量却常因"配体记忆(ligand memorization)"而精度不稳。本文提出的 PBCNet2.0 是一个基于笛卡尔张量的孪生等变图神经网络(E-GNN),在 860 万对蛋白–配体复合物上训练,于零样本(zero-shot)条件下在 FEP 基准上取得 Spearman ρ=0.67,与 FEP+(ρ=0.70)统计上无显著差异(P=0.39),同时保持高通量效率。文章进一步通过消融、可解释性与扰动实验论证了模型对几何约束的敏感性,揭示了其未经专门训练却涌现出的耐药突变预测能力,并在 ENPP1 与 ALDH1B1 两个真实靶点上完成了"预测→湿实验"的前瞻闭环,以 5/6 的命中率识别出功能性结合残基。本文从研究动机、架构数学、数据工程、基准评测、机理剖析到实验验证,对该工作做系统梳理与评析。
一、研究背景与动机
1.1 问题的位置
人类基因组中存在大量具有治疗潜力却仍被低估、未被充分研究的蛋白,其根源之一是缺乏高质量、被良好表征的分子探针。全球性的 Target 2035 计划即以"为所有人类蛋白找到药理学探针"为目标。在这一图景中,蛋白–配体结合亲和力预测是加速探针开发的关键环节。
当前的精度"金标准"是自由能微扰(FEP),如 Schrödinger FEP+ 在预测相对结合自由能上可达约 1.1 kcal/mol 的化学精度。但其高昂的计算成本、复杂的体系搭建与对专家干预的强依赖,严重限制了大规模应用。深度学习因此成为高通量亲和力预测的天然出口——直接从结构数据中学习,而非逐体系做物理模拟。
1.2 前作 PBCNet 的两点结构性局限
该团队此前提出的 PBCNet 是一个采用"成对比较"范式的孪生神经网络(SNN),其亲和力标签取自同一实验以最小化大规模数据库的系统误差,在高通量方法中已具优势,微调后甚至可逼近 FEP+。但作者明确指出两个限制:
- 1. 训练规模受限。 仅约 2 万个独特小分子、960 个靶点,化学与生物空间覆盖不足。规模定律(scaling law)表明,数据量与模型复杂度不足会直接制约泛化。
- 2. 几何编码僵化。 PBCNet 依赖原子对统计势(APSP)与固定角度分箱等预设几何特征,难以学习"随上下文而变"的相互作用,并对预设集合外的新原子类型表现不佳——这会妨碍对非常规结合模式的识别。
PBCNet2.0 的设计正是针对这两点:一边把数据"喂"够,一边把几何编码"放开",让模型从数据中直接学几何约束。
二、PBCNet2.0 的核心设计
2.1 整体框架
PBCNet2.0 沿用孪生架构,由三个模块串联构成:
模块 | 功能 |
|---|---|
消息传递模块(message-passing) | 在蛋白–配体相互作用图上传递信息,生成原子(节点)级表示 |
读出模块(readout) | 由节点表示聚合出复合物级与成对级表示 |
预测模块(prediction) | 三层前馈网络,将成对表示映射为相对亲和力 Δŷ |
推理时,模型只需两个结构相似的小分子(参照 + 查询)分别与同一靶点结合的构象,即可输出二者亲和力之差,从而对一个化合物系列做系统排序。其相对于前作的根本升级,在于消息传递模块改用基于笛卡尔张量、构建于 TensorNet 之上的等变 GNN。
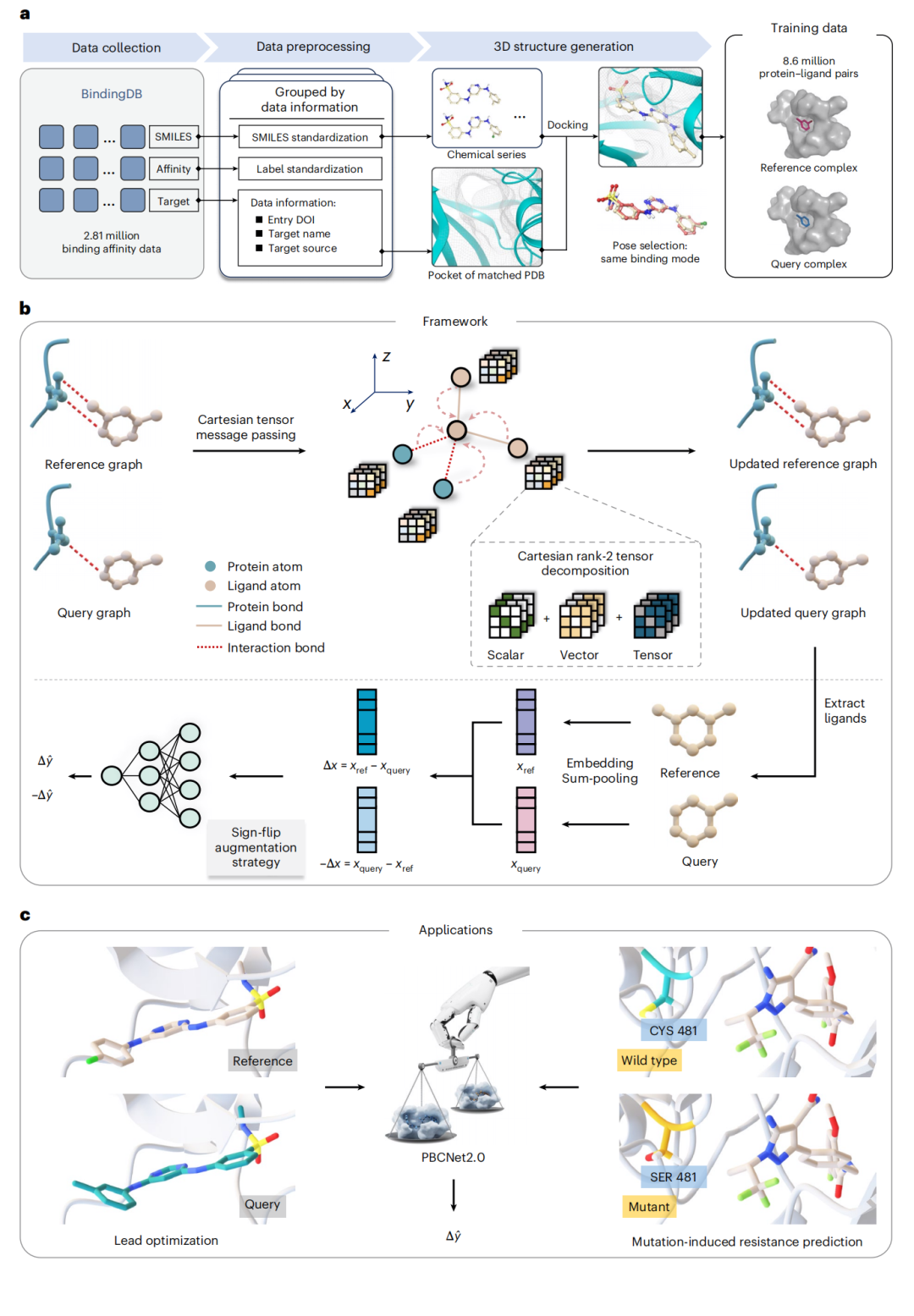

2.2 笛卡尔 rank-2 张量与不可约分解
PBCNet2.0 把每个节点的每一个隐藏维度编码为一个 3×3 的笛卡尔 rank-2 张量。任意张量 X 可做如下不可约分解:
X = (1/3)·Tr(X)·I + (1/2)·(X − Xᵀ) + (1/2)·(X + Xᵀ − (2/3)·Tr(X)·I)
↑ 标量(I) ↑ 矢量(A) ↑ 张量(S)
1 个自由度 3 个自由度 5 个自由度
旋转不变(scalar) 反对称→矢量变换 对称无迹→高阶张量- • 标量项正比于单位矩阵,旋转不变,1 个自由度;
- • 矢量项为反对称矩阵,3 个独立分量,按矢量方式变换;
- • 张量项为对称无迹矩阵,5 个独立分量,对应高阶张量。
这套表示的价值在于:三类特征通过 3×3 矩阵运算并行更新,在保持等变性(equivariance)的同时,能够同时编码距离与角度等三维几何信息。相比标量型 GNN 只能感知"远近",张量表示天然携带"方向/角度"——这正是后文识别氟正交多极相互作用所需的能力基础。
设计要点:对 TensorNet 的关键改动。 原始 TensorNet 在边的初始化中含有一个距离衰减函数,用于抑制长程相互作用,但会在相关性权重中引入距离依赖的偏置。PBCNet2.0 显式移除了该函数(在初始张量构建与消息计算两处均移除),使模型不再人为压低长程项,而由数据自行决定相互作用的距离尺度。这是其"几何放开"理念的具体落点。
2.3 图构建与特征初始化
- • 口袋定义: 取配体 8 Å 范围内的残基为蛋白口袋。每个重原子为节点,共价键为边。
- • 距离边: 蛋白–配体、配体–配体节点对在空间距离 ≤5.0 Å 时建边。
- • 蛋白–蛋白对的双阈值: 若某蛋白原子已通过距离边连到配体原子,则其与另一蛋白原子在 ≤5 Å 时建边;否则采用更严格的 ≤3 Å。该分层策略优先保留蛋白–配体相互作用区域,同时削减长程蛋白内连接以降低计算复杂度。
- • 特征: 在 TensorNet 基础上扩展为 9 类原子特征与多种键类型,以更完整地刻画原子及其局部化学环境。边的距离信息经**径向基函数(RBF)**展开后,与原子/键的化学特征一起,通过线性层编码进初始张量。
2.4 消息传递与等变更新
模型堆叠 3 层消息传递。每层中,节点张量先做 Frobenius 范数归一化,再分解为 I/A/S 三部分并经独立线性组合更新;随后将"键不变嵌入 + 两端原子的 O(3) 不变标量嵌入"融合,得到三个不变门控因子 f_I、f_A、f_S,用以加权来自邻居的消息 M_ij。消息聚合后,以**保宇称(parity-preserving)**的形式 X·M + M·X 更新张量,再经归一化与残差连接得到下一层表示。整个流程严格维持等变性,确保几何信息在传播中不被破坏。
2.5 读出层:为何只用差向量 Δx
这是 PBCNet2.0 一个看似细微、实则关键的设计选择。
模型对配体原子的(O(3) 不变)标量表示做池化,得到参照复合物表示 x_ref 与查询复合物表示 x_query。成对表示仅取差向量:
Δx = x_ref − x_query而非 PBCNet 中 [x_ref, x_query, Δx] 的拼接。原因在于:拼接方案下,网络原则上可以主要依赖两个绝对表示 x_ref、x_query 取得不错的训练表现,而在很大程度上忽略差值项——这会鼓励"配体特异性记忆",而非学习"结构变化如何转化为活性变化"。仅用 Δx,则迫使模型直接把两个复合物之间的结构差异映射到相对亲和力,与成对学习目标对齐,从而缓解配体记忆。
作者给出了直接的经验证据:在 FEP 测试集上,重新实现的拼接方案 [x_ref, x_query, Δx] 仅得 ρ=0.59,而仅用 Δx 的 PBCNet2.0 为 ρ=0.67。
2.6 反对称约束与符号翻转增强
理想的相对亲和力函数应满足反对称性:f(Δx) = −f(−Δx)(交换参照与查询,差值应反号)。为此,训练时同时输入 −Δx = x_query − x_ref 并联合计算损失,作为一种计算开销极低的数据增强(符号翻转/标签翻转)。损失采用对每一对样本的两个朝向——原始 (ref, query, y) 与翻转 (query, ref, −y)——平方误差取平均后开方的 RMSE 形式,使反对称约束在训练中显式化。
2.7 训练数据管线
8.6M 训练对来自一条三阶段流水线:
- 1. 数据采集。 从 BindingDB(2023.12 版)提取约 281 万条亲和力测量(IC₅₀、EC₅₀、Ki、Kd)。
- 2. 预处理与质控。
- • 剔除 RDKit 无法解析的非法 SMILES;剔除标为 NaN、0 或含不等号(
</>)的条目。 - • 按 entry DOI、靶点名、靶点来源分组,保证实验上下文一致;组内以 Tanimoto 0.4 阈值聚成化学系列。
- • 同一分子多值时:若最大/最小亲和力之比 < 3 倍取算术均值,否则剔除。
- • 为每个系列选取与系列配体 Tanimoto 相似度最高的 PDB 结构作对接模板;无共晶配体相似度 >0.5 的系列被剔除。
- • 剔除 RDKit 无法解析的非法 SMILES;剔除标为 NaN、0 或含不等号(
- 3. 三维结构生成。 用 Glide 对接(LigPrep 备配体、Protein Preparation Wizard 备蛋白),保留与配体/受体形成 >3 个氢键的保守水;每个配体最多生成 200 个构象。位姿筛选用最大公共子结构(MCS)对齐,保留 MCS 区域内 RMSD ≤ 2.0 Å 的位姿,多位姿时取 Glide 打分最高者。
- 4. 配对与分布平衡。 每个系列最多配对 15,000 对,优先纳入此前未配对的配体。原始标签呈集中于 [−1, 1] 的正态分布,存在让模型退化为"预测均值附近"的过拟合风险;为此对 |ΔpAct|≥1 区域欠采样、对 |ΔpAct|<1 区域过采样,使分布更均匀。最后剔除与测试集重叠的对,确保数据独立性。
升级后训练集覆盖 0.28M 独特小分子、1,122 个靶点,较前作在靶点覆盖与化学多样性上均显著提升。
三、性能评测
3.1 评测设置
基线(12 种,四类):
类别 | 方法 |
|---|---|
传统物理/打分 | Glide SP、MM-GB/SA、Schrödinger FEP+ |
共折叠(co-folding) | Boltz-2 |
序列深度学习 | PSICHIC、BIND、PLAPT |
结构深度学习 | LigUnity、PIGNet2、RTMScore、GenScore、OnionNet-2、PBCNet |
测试集:
测试集 | 构成 | 考察重点 |
|---|---|---|
FEP | FEP1(8 系列,含 FEP+ 预测)+ FEP2(8 同系物系列)= 16 系统 | 零样本相对亲和力排序 |
SAR-Diff | 8 对(共 16)化学系列,同 R 基改造但 SAR 不同(源自 ChEMBL) | 解耦结构改动与活性变化 |
Mutation | 8 个靶点(源自 MdrDB),仅保留保持相互作用模式的口袋突变 | 突变诱导的亲和力变化 |
F-Opt | 10 对复合物,-H/-CH₃ → -F/-CF₃ | 氟正交多极相互作用 |
指标: Pearson r、Spearman ρ、RMSE、MAE。FEP 自由能经非竞争结合假设换算为 pIC₅₀(297 K)。
3.2 零样本:逼近 FEP+ 这一金标准
在 16 个化学系列上,PBCNet2.0 取得全场最佳预测能力。
方法 | Spearman ρ | Pearson r |
|---|---|---|
Schrödinger FEP+ | 0.70 | 0.70 |
PBCNet2.0 | 0.67 | 0.66 |
Boltz-2 | 0.58 | — |
PBCNet(前作) | 0.57 | 0.56 |
GenScore | 0.45 | — |
MM-GB/SA | 0.40 | — |
- • 相对前作提升 18%(ρ 0.57→0.67);
- • 与 FEP+ 的差距仅 Δρ=0.03、Δr=0.04,双侧 Wilcoxon 检验显示二者无显著差异(P=0.39)——一个高通量模型在关键排序指标上追平了工业级物理模拟;
- • 对其余多数基线的优势具统计显著性(多数 P<0.01)。
预测值范围也更"贴合实验":在 pfkfb3 系统中,PBCNet2.0 预测跨度为 [−2, 2],与实验范围吻合,而 PBCNet 仅限于 [−1, 1]。
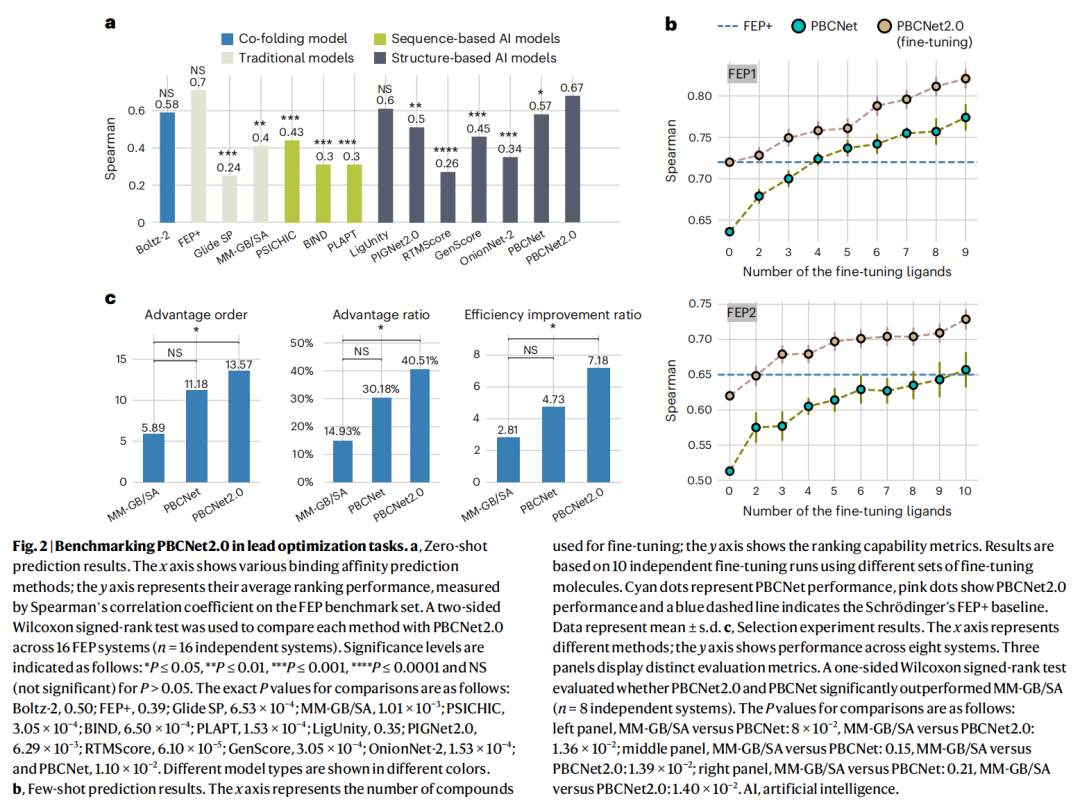
3.3 少样本微调
仅用少量已知活性分子做微调,PBCNet2.0 性能随微调配体数稳步攀升,持续超越前作甚至 FEP+ 基线。这种数据高效的精炼能力,使其在"某靶点仅有零星 SAR 数据"时尤为实用。
3.4 优先级排序实验(8 个药学靶点)
这是最贴近工业优化场景的回顾性评测,三项指标全面领先:
指标 | MM-GB/SA | PBCNet | PBCNet2.0 |
|---|---|---|---|
优势序(advantage order) | 5.89 | 11.18 | 13.57 |
资源节省率(advantage ratio) | 14.93% | 30.18% | 40.51% |
效率提升倍数(vs 随机) | 2.81× | 4.73× | 7.18× |
效率提升 7.18 倍对应约 717.81% 的相对改进;统计检验确认 PBCNet2.0 显著优于 MM-GB/SA(P≤0.05)。在采用相同成对/排序范式但架构不同的 LigUnity 之上,PBCNet2.0 在 FEP 基准上仍占优。
3.5 相似度敏感性
在 FEP 测试集上,模型精度随参照–查询相似度优雅退化:
- • 高相似对(ECFP4 Tanimoto ≥ 0.8):RMSE ≈ 0.80 kcal/mol;
- • 中相似对(0.6–0.8):仍保持强表现;
- • 结构差异较大对(<0.6):误差平滑上升,但仍约 1.1 kcal/mol。
说明模型在较宽的系列内相似度范围内都稳健,而非仅对"近邻类似物"有效。
四、机理剖析:模型究竟学到了什么
机器学习打分模型的通病是"预测主要由配体记忆而非相互作用理解驱动"。作者用三组实验正面回应了这一质疑。
4.1 消融实验:相互作用信息的主导地位
将分子图中所有蛋白–配体相互作用边移除,得到"残缺图",与"完整图"对照监测训练动态:
- • 初期(约 12,500 步内): 两条曲线趋势相近——模型初期主要依赖分子结构信息;
- • 越过该点后:残缺图性能持续下滑,完整图持续上升。
这表明蛋白–配体相互作用信息已成为预测的主要决定因素,模型在训练中逐步发展出对相互作用的理解。
4.2 SAR-Diff:把结构与活性解耦
SAR-Diff 专门设计为:同一 R 基改造,在配对的两个系列中却对应完全不同的 SAR 与核心骨架,且系列间活性无相关。要在此取得成功,模型必须分析结合口袋的相互作用,而不能靠结构模式记忆。
结果:PBCNet2.0 在全部 8 对系列上均具一致预测力,而 Boltz-2 仅在 4 对成功;PBCNet2.0 与 PBCNet 是唯二从未在任何系列对上完全失败的方法。在平均 ρ 与 RMSE 两项上 PBCNet2.0 均最优。值得注意的是,对多数模型而言 SAR-Diff 比 FEP 更具挑战性,进一步凸显其稳健性。
4.3 氟正交多极相互作用:原子级洞察的试金石
氟正交多极相互作用是一种重要却常被忽视的相互作用:氟的高电负性形成亲核中心,羰基氧的吸电子效应使羰基碳亲电,二者电子互补。但它有严格几何要求——F–C 距离须在 3.0–3.7 Å、F···C=O 夹角接近 90°,连许多通用力场都难以准确刻画。准确识别它,需要同时精确建模三维空间关系与角度依赖。
在 10 对样本的 F-Opt 基准上:
方法 | MAE | 符号一致性 |
|---|---|---|
PBCNet2.0 | 0.33 | 100% |
PBCNet | 0.52 | 100% |
其他方法 | 更高 | 错误率 20%–50% |
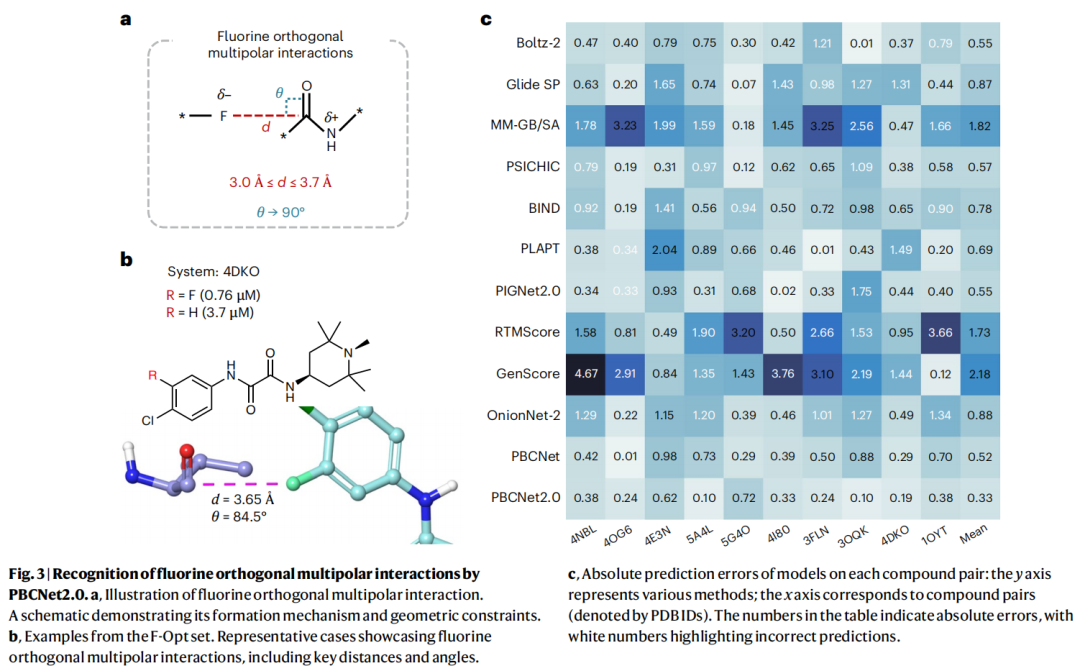
仅 PBCNet2.0 与 PBCNet 实现 100% 符号一致(从不把"变强"判成"变弱")。
更关键的是角度敏感性的因果证据:在 5G4O 与 4E3N 体系上,模型给参与相互作用的碳原子赋予 0.98–1.00 的高权重;随后用 Maestro 做扰动——将两个关键夹角从 79.2°→52.9°、95.6°→133.7°(均推出合理范围),碳原子权重随之跌至 0.50 与 0.56。这直接证明:等变框架使模型真正对角度约束敏感,而非凭统计偏好"蒙对"。作为对照,前作 PBCNet 的 APSP 对"氟–羰基碳"在相关距离上赋予了偏低的相互作用概率,且对角度参数敏感性有限——这正是其三维建模较粗糙的体现。
4.4 可解释性方法
文中可视化的"权重",是从消息传递第一层提取的归一化标量门控因子。具体地,取该层三个不变项的平均:
f_ij = (f_I + f_A + f_S) / 3再对中心原子所有邻居做 min–max 归一化得到 0–1 的权重 w_ij,用以量化邻居原子对中心原子的相对影响。一个理想模型会倾向于给"可能形成关键相互作用的原子对"更高权重,因为它们对彼此化学环境及结合亲和力影响更大。氢原子因依赖于程序的定位与高计算开销被排除;对氢键供体,则取与氢共价相连的重原子参与分析。
五、涌现能力:预测耐药突变
这是本文最具想象力的部分。
5.1 任务迁移假设
残基突变可改变口袋性质与结合亲和力,进而导致耐药。NSCLC 中第一代 EGFR 抑制剂(如吉非替尼)治疗后获得性 T790M 突变在 9–14 个月内出现于 >50% 耐药病例,通过空间位阻阻碍药物结合,催生了第三代抑制剂奥希替尼。因此,提前预测可能影响配体结合的突变对优化与探针开发意义重大。
PBCNet2.0 从未在突变数据上训练(训练集仅含"不同配体结合同一蛋白位点")。但作者假设:既然"换配体"与"换残基"在本质上都是在改变蛋白–配体相互作用,前者的能力或许可迁移到后者——这与模型已展现的"捕捉微妙相互作用"能力一脉相承。
5.2 突变基准结果
在含 8 个临床相关靶点(均带口袋突变)的 Mutation Benchmark 上:
- • PBCNet2.0 平均 ρ = 0.53;
- • Boltz-2、Glide SP、BIND、PLAPT、RTMScore、GenScore 等甚至出现负相关——基本完全失败。Boltz-2 失效或源于共折叠方法难以准确预测序列突变引起的结构变化,导致其亲和力模块崩溃。
靶点级别上表现分化:tACE(ρ=0.76)、HIV-1 蛋白酶(ρ=0.75)突出;AKR1B1(ρ=0.28)、PfDHFR-TS(ρ=0.26)则欠佳。
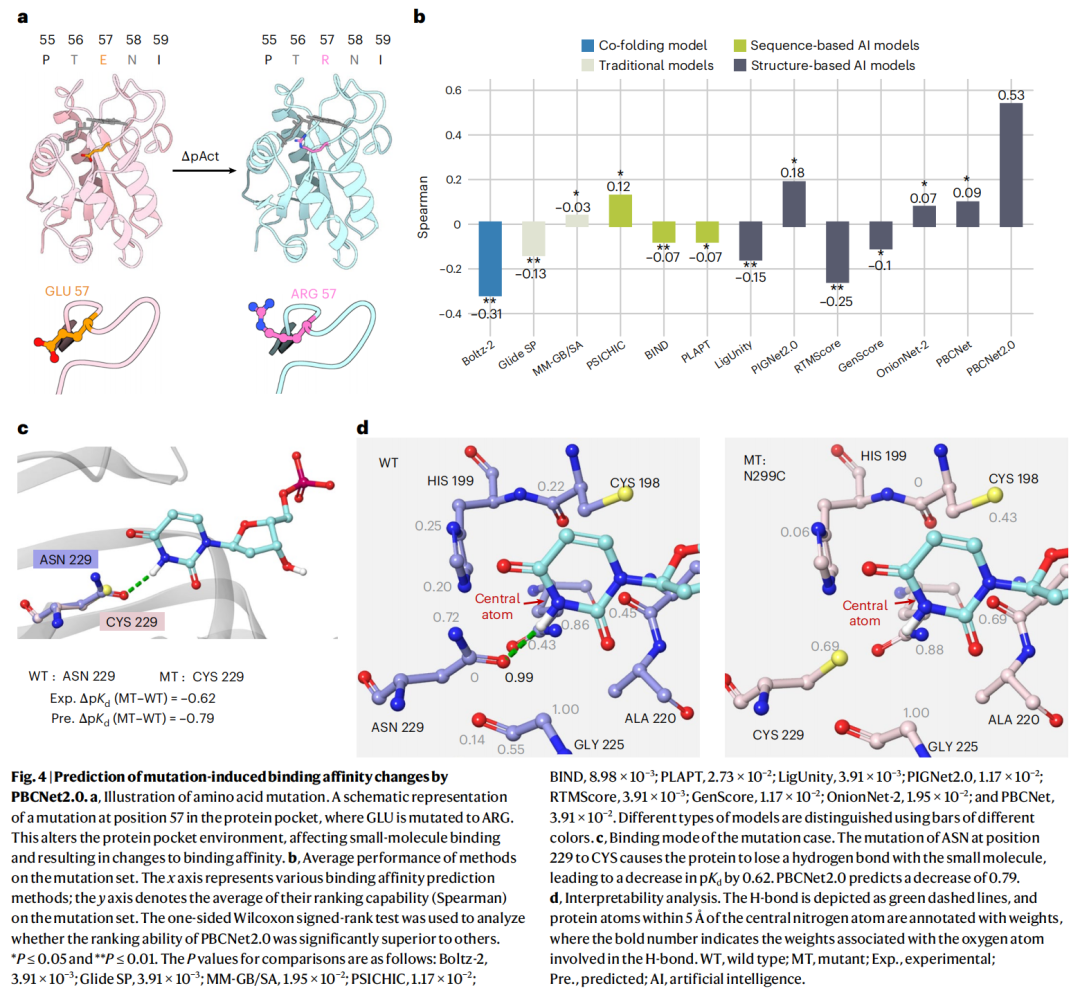
5.3 机理验证与回顾性案例
机理可解释。 在 Lactobacillus casei 胸苷酸合成酶(LTS)上,N229C 突变破坏了 ASN229 酰胺氧与配体吡啶氮之间的关键氢键,实验测得亲和力下降 ΔpKd = −0.62,模型预测 −0.79,高度吻合。可解释性显示:野生型中该参与氢键的氧原子被赋予 0.99 高权重,突变后信号完全消失——模型不仅"算对"还"指对了原因"。
回顾性案例。 对三款突变选择性抑制剂,模型在结构迥异的体系上均给出准确的相对亲和力:
- • olutasidenib(靶向突变型 IDH1,2022 年 12 月 FDA 批准,约 1,000 倍选择性);
- • MRTX1133(KRAS^G12D 非共价抑制剂,2023 年 3 月进入临床);
- • pirtobrutinib(首个非共价 BTK 抑制剂,已获 FDA 批准)。
这种涌现能力,作者认为源于模型在 860 万级数据上习得的对蛋白–配体识别的通用理解——正如大模型在规模足够后"突然学会"未被显式训练的技能。
六、前瞻性实验验证
回顾性结果再好,也比不上"先预测、后实验"的前瞻闭环。作者在两个真实靶点上完成了全套湿实验。
6.1 ENPP1:验证氟相互作用是否真实存在
基于已知抑制剂 A1(R=F),设计仅将氟换为氢的类似物 A2(R=H),以在最小结构扰动下消除潜在偶极效应。可解释性显示 A1 的氟与靶点羰基氧之间的潜在偶极相互作用权重高达 0.94;以 A1 为参照,模型预测 A2 结合力骤降约 40 倍(ΔpAct = 1.61)。
实验 | A1(R=F) | A2(R=H) |
|---|---|---|
热迁移 ΔTm | +3.8 °C | +2.7 °C |
酶活 IC₅₀ | 13.3 nM | 2,284 nM |
SPR Kd | 35.3 nM | 1,030 nM |
SPR 实测 ΔpKd = 1.46,与模型预测 1.61 仅差 0.15。多套正交实验一致,有力证实了 A1 与 ENPP1 之间稳健的氟介导正交多极相互作用。
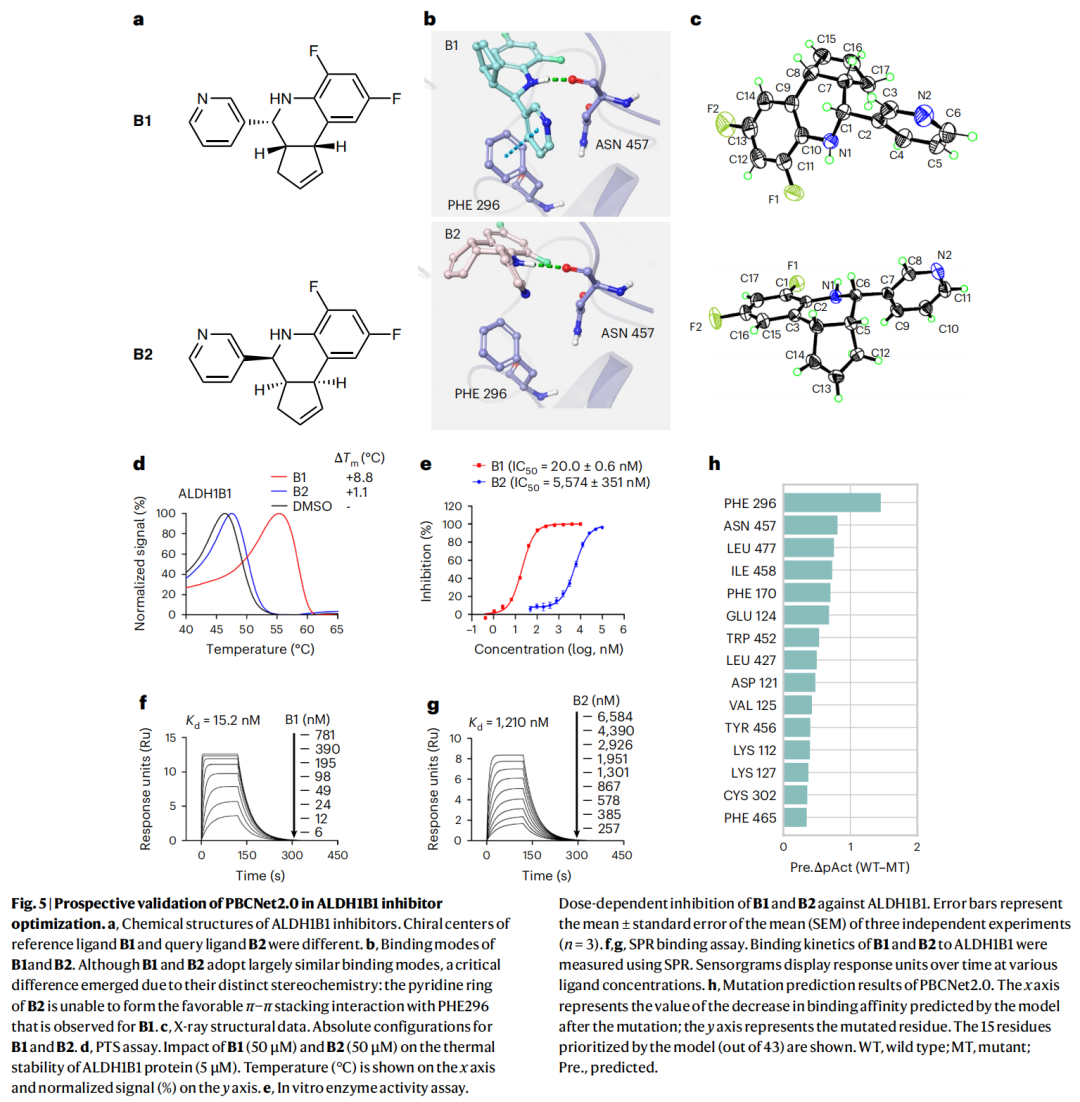
6.2 ALDH1B1:手性识别 + 耐药残基定位
① 区分对映体。 对映异构体 B1 / B2 中,分子对接显示二者结合模式总体相似,但 B2 的吡啶环因立体化学差异无法与 PHE296 形成有利的 π–π 堆积。模型预测 B1 活性约为 B2 的 10 倍(ΔpAct = 1.07),可解释性也凸显了 B1 吡啶环与 PHE296 的 π–π 堆积。
实验 | B1 | B2 |
|---|---|---|
热迁移 ΔTm | +8.8 °C | +1.1 °C |
酶活 IC₅₀ | 20 nM | 5,574 nM |
SPR Kd | 15.2 nM | 1,210 nM |
X 射线晶体学确定了二者绝对构型,实验亲和力差异与预测一致,证明模型能捕捉立体化学依赖的活性差异。
② 定位耐药残基。 通过计算丙氨酸扫描逐个把口袋残基突变为丙氨酸,用模型预测每个突变体–B1 复合物相对野生型的亲和力变化,据此挑出 6 个残基做实验验证。site-directed mutagenesis 配合 PTS 与 SPR 显示,其中 **5 个(PHE296、ASN457、LEU477、ILE458、PHE170)**的丙氨酸取代显著削弱 B1 结合,仅 GLU124 例外——命中率 5/6。作为对照,传统 MM-GB/SA 虽将 PHE296、PHE170 排在前列,却把 ASN457、ILE458、LEU477 排到很靠后的位置。
三点小结:模型在此实现了**(1)** 经对接+可解释分析的准确结合模式预测、(2) 识别出比外消旋前体更优的纯手性抑制剂 B1、(3) 经计算丙氨酸扫描成功预测潜在耐药突变。
七、局限性与未来方向
作者对适用边界保持了难得的坦诚:
- • 静态构象的固有约束。 模型依赖静态结合构象,无法显式考虑结合过程中的熵效应与构象动力学。结合蛋白–配体共折叠方法引入蛋白柔性,是重要的未来方向。
- • 结合模式改变 / 完全失活难题。 当 R 基改造导致结合模式整体改变时预测困难。这源于公共库中经验证的阴性数据稀缺,以及测定极低活性化合物的技术困难;因 PBCNet2.0 比较的是"结合模式相近"的化合物,事先做位姿验证可过滤掉结合模式被破坏的分子。
- • 定位:优化而非"大海捞针"。 由于 BindingDB 等公共活性库结构性偏向高亲和力配体、不系统报告真正无活性分子,PBCNet2.0 主要面向从中/高活性苗头出发、且有合理结合位姿"的先导与探针优化,而非在大量无活性分子中做普适的苗头发现。
- • 依赖结构锚点。 适用于至少有一个可靠共晶结构或高置信锚定位姿、且保持合理结合模式的同系物系列;扩展到缺乏结构锚点的靶点与化学型,需待共折叠与全原子预测的位姿精度足够稳健。
未来方向:(1)将高质量突变数据纳入训练以增强相互作用刻画与跨靶点泛化;(2)将其训练框架用作预训练策略,微调到虚拟筛选等下游任务,缓解传统方法的配体记忆偏差;(3)继续扩大规模——在已探索范围内,模型在训练算力、模型规模与数据规模上均呈幂律式增长且尚未饱和,继续加数据、加参数有望进一步提升。
工程友好性: 整个模型仅在单张 NVIDIA A100(80 GB)上训练约 7 天,为复现与进一步扩展提供了务实参考;代码与测试数据均以 MIT 协议开源,训练数据托管于 Zenodo。
八、总结与评价
PBCNet2.0 在三个层面给出了有分量的推进:
- 1. 方法层面——把"几何"真正交给数据。 用笛卡尔张量等变表示替代预设势函数,并移除 TensorNet 的距离衰减偏置,使模型能从数据中学到距离与角度约束;仅用差向量 Δx 的读出设计,从机制上抑制了配体记忆。这些选择都直接对应到可量化的收益(如 Δx vs 拼接的 0.67 vs 0.59)。
- 2. 能力层面——精度、可解释与涌现三者兼得。 零样本追平 FEP+(P=0.39)、对氟相互作用的角度敏感性有因果证据、以及未经训练即可迁移到耐药突变预测,共同说明模型获得的不是"表面拟合",而是对蛋白–配体识别较为通用的理解。
- 3. 证据层面——真前瞻、真湿实验。 ENPP1 的 ΔpKd 预测误差仅 0.15、ALDH1B1 的 5/6 残基命中,把"纸面 SOTA"落成了可信的实战工具。
值得读的理由可归纳为四点:(1) "又准又快"从口号变为可统计验证的结论;(2) 模型判断可与物理化学原理逐一对应,不是黑箱;(3) "涌现"出的耐药预测能力本身极具启发性,为耐药分析开辟新路径;(4) 预测–实验闭环完整,工程门槛友好、代码开源。
需要清醒看待的是其静态构象与面向优化而非发现的定位——它不是"端到端从零找苗头"的万能解,而是一把在已有活性苗头与结构锚点之上、做高效优化与耐药研判的利器。在当下蛋白–配体共折叠快速演进的背景下,如何把柔性与动力学接入这套等变框架,将决定它能否从"优化助手"进一步走向"发现引擎"。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-16,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号