Nat. Rev. Phys. | 数据驱动的生物动力学模型发现

Nat. Rev. Phys. | 数据驱动的生物动力学模型发现

DrugAI
发布于 2026-06-29 13:43:05
发布于 2026-06-29 13:43:05
动力系统理论为描述相互作用的生物组分如何随时间和空间演化提供了数学框架,适用范围从分子振荡器到大尺度生物模式。许多生物系统包含非线性反馈、时间延迟和多尺度相互作用。随着实验测量变得更加丰富且维度更高,传统机制模型的构建也变得越来越困难。这推动了数据驱动方法的发展:这些方法直接从数据中推断模型结构,为构建动力学模型提供了机制建模之外的替代路径。
在这篇技术综述中,研究人员讨论并比较了用于发现生物动力系统模型的数据驱动方法,重点关注三类主要方法:基于回归的方法、基于网络的架构以及分解技术。

生物系统在多个尺度上表现出丰富的动力学行为。纯时间动力学的例子包括控制细胞分裂的细胞周期振荡器、使生理活动与昼夜节律同步的生物钟、调控受精和神经信号传导的细胞内钙振荡、酵母代谢中的糖酵解振荡、DNA损伤反应中的p53振荡,以及由阈值触发离子电流产生动作电位的神经兴奋性。时空动力学的例子则包括早期胚胎发育中的有丝分裂波、社会性变形虫聚集过程中的环腺苷酸波,以及再生组织中的Erk活性行波。在这些系统中,非线性反馈、时间延迟和空间耦合共同产生了复杂的时间和空间模式。
为了在可处理的条件下研究这些现象,研究人员通常会构建简化系统,使其能够捕捉相同的基本动力学原理。动力系统的数学模型为描述和分析这些系统提供了一种统一语言。一个动力系统可以理解为:一组状态变量在既定规则和参数控制下随时间演化。理解一个动力系统通常包含多个互补目标,即根据当前测量预测未来状态,识别导致观测行为的变量及反馈网络,并刻画系统可能出现的解,例如稳态、极限环以及它们之间的转变。
从原则上说,如果能够推断完整的控制方程,就可以同时解决这些问题。但在实践中,对于高维、观测不完全且相互作用不确定的生物系统,完整推断通常不可行。因此,现代数据驱动方法试图直接从时间分辨数据中重构系统动力学的一个或多个方面,从而在机制建模与经验推断之间建立桥梁。
传统上,动力系统研究主要依赖基于方程的建模,即从第一性原理或机制直觉出发推导控制规律,例如牛顿定律或化学振荡的动力学模型。对于低维且机制明确的系统,这种方法依然非常有力,但对于大型生物网络则会迅速变得难以处理。实验测量和计算能力的进步,使一种互补范式成为可能:直接从数据中推断动力学结构。机器学习和统计方法,例如非线性时间序列重构或现代神经网络模型发现,现在可以在没有完全指定机制模型的情况下提取预测结构。在生物学中,这些技术越来越多地应用于活细胞成像、单细胞动力学和组学时间序列。不过,单纯的预测准确性并不足够;要从复杂生物数据中获得因果理解,还需要机制约束。
在这篇技术综述中,研究人员聚焦于从时间分辨生物数据中学习复杂系统控制动力学的方法,并以振荡过程作为非线性生物行为的典型例子。尽管许多生物系统由空间扩展模型控制,本文重点并不是偏微分方程的识别,而是从时间分辨数据中发现有效的低维动力学模型,例如常微分方程和潜在状态表示。研究人员比较了代表性的基于回归、基于网络和基于分解的方法,并使用Oregonator模型作为统一示例。该模型能够捕捉基本的非线性振荡行为,同时在解析和计算上仍然可处理。
研究人员希望提供一个可用工具的概览,说明不同方法的适用场景,并展望自然科学中数据驱动模型发现的未来方向。本文的目标读者不仅包括物理学家,也包括化学家和生物学家。研究人员主要关注确定性方法,而不讨论模拟推断、近似贝叶斯计算和贝叶斯模型选择等概率框架。尤其强调那些关注动力学结构、可解释性以及与Koopman理论相联系的互补方法。
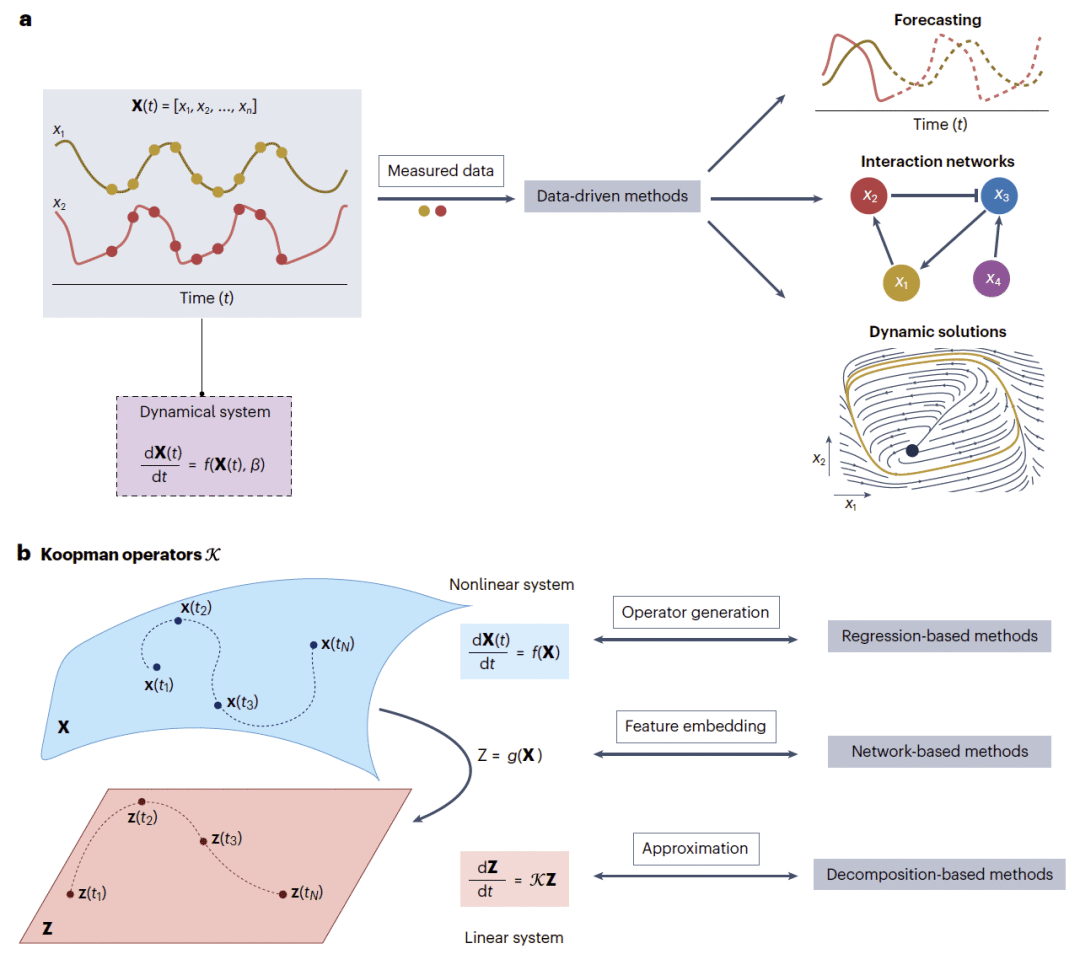
图1|动力系统的数据驱动方法与Koopman算子框架。
Koopman算子提供统一框架
从线性系统到算子思维
在线性动力系统中,分析相对简单,因为系统可以被显式求解并分解为彼此独立的组成部分,即模态。通过变换到特征向量坐标,每个模态可以独立演化并单独分析。特征值的实部决定系统稳定或不稳定,虚部决定振荡频率。
大多数生物系统是非线性的,不能像线性系统一样直接对角化。不过,Koopman算子理论提供了一个统一视角:与其直接分析原始非线性状态变量,不如将它们转换为一组新的函数,也就是“可观测量”。在这些可观测量构成的空间中,动力学可以变为线性。若有限个可观测量能够捕捉所有相关动力学,就可以像分析线性系统一样进行特征分析。对数据驱动方法而言,核心挑战就是如何从数据中自动发现这些合适的可观测量。
从算子思维到方法分类
Koopman理论在经典线性分析和现代数据驱动方法之间建立了概念桥梁。许多研究非线性动力系统的方法都可以被解释为以不同策略近似Koopman算子的作用,不论这种近似是显式的、隐式的,还是局限于某些可观测量子空间中的。
本文按照方法学基础将数据驱动方法分为三类。第一类是基于回归的方法,它们通过回归检验符号模型方程或变量之间推断出的连接是否能够解释观测数据。与预先指定方程的经典建模不同,这类方法在拟合过程中选择或修正方程。它们通常具有符号形式,因而有助于解释,但在高维场景中容易受到维度灾难影响。第二类是基于网络的方法,例如神经网络架构,它们用于捕捉复杂非线性关系。这类方法通常在高维潜在空间中学习合适的可观测量以近似Koopman算子,往往能够获得较好的预测能力,但直接可解释性可能受限。第三类是分解方法,它们通过数学变换直接从数据中提取主导时空模态,而不需要显式控制方程。许多模态对应于Koopman模态,使复杂非线性系统能够被表示为低维或线性结构。
这种按照方法学而不是按照“监督/无监督”或“白箱/黑箱”分类的方式,是有意为之。白箱和黑箱标签本身并不能完全决定模型是否可解释。例如,稀疏回归模型通常被视为白箱,但若应用于高维、噪声强且基函数选择不当的数据,也可能产生难以解释的结果。相反,神经网络通常被称为黑箱,但如果通过架构约束或加入物理信息特征,也可以提升解释性。因此,关注具体方法而不是哲学标签,更有助于理解各类方法在预测、相互作用推断和动态解识别中的优势与局限。
尽管数据驱动建模领域非常广泛,新方法的评估通常围绕少数典型动力系统展开,例如低维振荡器、混沌系统以及描述输运或湍流的偏微分方程。由于不同方法的目标差异很大,很难用一个统一数值基准评价所有标准。用于符号发现的方法优先考虑简约性和结构真实性,而水库计算等方法则更侧重复杂或混沌动力学的高保真预测。因此,某种分解方法在预测方面评价较弱,并不意味着算法失败,而可能反映一种设计取舍:它更关注潜在状态识别或动力学结构,而不是长期时间外推。
基于回归的方法
基于回归的方法是识别动力系统中最常用的数据驱动工具之一。它们试图通过显式函数关系推断一个测量变量的变化如何由其他变量解释。一般而言,回归任务就是选择一个模型结构和参数,使其尽可能准确地预测观测数据。
用于动力系统模型发现的基于回归方法大体可分为三类:强调因果推断的方法、基于预定义候选库和稀疏回归的方法,以及采用进化算法的方法。尽管方法学不同,这些方法有两个共同特征。首先,它们通常产生符号化、可解释的模型,可以表现为相互作用网络,也可以表现为显式微分方程。其次,它们本身无法判断数据中是否包含了所有相关状态变量,也就是说,无法确认观测变量是否真正覆盖了系统底层状态空间。这些优势和局限共同决定了它们在生物学中的应用方式。
因果方法
因果方法旨在推断一个变量是否有助于解释另一个变量的未来。在生物学中,这类推断常用于从时间序列中重构相互作用网络,例如基因表达动力学或生态动力学中的网络结构。
经典方法之一是Granger因果,它通过比较是否加入另一个变量过去值对预测准确性的影响来判断因果关系。若加入某变量的历史信息后,目标变量的预测误差下降,则该变量被认为对目标变量具有Granger因果作用。这一方法简单且应用广泛,但在非线性、振荡或同步耦合系统中表现较差,容易产生虚假的全连接网络。
也有一些基于约束的替代方法可以缓解这些问题。例如,一般常微分方程推断方法会检验观测时间序列是否与符号一致的调控效应相容,并利用时间差异和导数差异统计量剪除假阳性,同时识别更高阶调控模式。
从Koopman视角看,基于回归和基于约束的因果推断方法都可以看作是在比较不同可观测量集合对系统演化的捕捉能力。在Granger类型方法中,这种比较是显式的:研究人员比较仅由目标变量历史构建的预测器与同时包含其他变量历史的预测器。在一般常微分方程推断中,这种比较则是隐式的:加入或移除候选调控因子会改变用于测试系统演化结构的可观测量集合,因果性则由这些结构是否满足假定方程形式中的符号一致性约束来推断。
预定义库的稀疏回归
稀疏回归方法通过从预定义候选函数库中选择少量活跃项来重构控制方程。这类方法的关键在于预先指定函数基,而不是限制为某一种具体函数形式。代表性方法包括带外源输入的非线性自回归滑动平均模型,以及非线性动力学稀疏识别方法SINDy。
NARMAX最初来自控制理论,用预定义的函数展开描述离散时间动力学。它可以包含过去状态、外源输入和残差项,并可使用多项式、有理函数或径向基函数等不同展开形式。NARMAX通常更强调预测准确性和可控性,而不是解释性,因此已被用于生物力学、生理学和生化信号预测等领域。
与固定多项式回归不同,SINDy强调简约性和可解释性。它在用户定义的候选函数库上执行稀疏回归,识别最少数量的活跃项。函数库并不局限于多项式,也可以包括有理函数、三角函数、指数项或其他与问题相关的基函数。该方法假设真实动力学是稀疏的,也就是说,大多数候选项系数为零,仅有少量项真正参与系统演化。从Koopman角度看,SINDy可以被理解为用候选函数作为提升函数,近似Koopman生成元。
这类基于函数库的稀疏回归方法具有良好可解释性,尤其当领域知识能够指导基函数选择时非常强大。但它们仍然依赖预定义函数库,在高度非线性的生物系统中可能限制发现能力。另一个局限是对数据质量高度敏感。由于回归通常需要估计导数,噪声或时间分辨率不足会严重扭曲结果。虽然已有改进的微分估计方法和抗噪扩展方法,但在真实生物数据中,有限观测、噪声测量和不完整先验知识仍会使SINDy迅速失效。
进化方法
进化算法,尤其是符号回归,可以突破预定义函数库对未知动力学捕捉能力的限制,因为它能够同时发现候选模型的结构和参数。在符号回归中,模型被编码为层级表达式树,内部节点代表运算符,叶节点代表变量或常数。这些树通过多代进化逐渐优化,主要操作包括突变、重组和选择。突变会用随机生成的新子树替换原有子树,重组会在两个父代模型之间交换子树,选择则保留预测误差更低或更简约的模型。这种进化搜索允许在不依赖固定函数库的情况下发现新结构和参数值。
符号回归非常灵活,但也计算成本高,容易过拟合,并且由于搜索空间巨大而对噪声敏感。这些问题可以通过加入领域约束来缓解。AI-Feynman就是一个典型例子,它利用物理先验,如量纲一致性、简约性、组合性、对称性和可分离性来限制搜索空间。这说明,精心选择约束既可以加快收敛,也可以增强解释性。
在生物学中,类似物理学那样的通用规则更难定义,但仍可借助生化先验、守恒定律或已知调控模体来加入约束。符号回归已经被应用于基因调控和生化网络、生态动力学、细胞相互作用网络和机制性组织模型等领域。不过,这类方法的成功通常依赖系统特异性知识来约束搜索。概念上,符号回归可以被看作一种非常灵活但结构相对松散的Koopman提升函数近似方式,它用计算效率换取通用性。当受到合适先验引导时,进化方法在复杂生物系统的可解释模型发现中具有重要潜力。
生物学应用中的局限
在不同子类中,基于回归的方法面临三个反复出现的问题。第一是严格的数据要求。依赖显式数值微分的方法会放大噪声,因此通常需要密集且高时间分辨率的时间序列,但生物数据常常无法满足这些条件。第二是对先验知识的依赖。方法成功依赖是否能够观测所有相关状态变量,以及函数库或算子集合是否选择合理。缺少这些先验时,模型可能变得虚假或不可解释。第三是尺度扩展问题。高维生物网络会加剧维度灾难,限制方法适用性。
近年来,弱形式和积分形式的稀疏回归方法部分缓解了噪声放大问题。这些方法避免显式数值微分,而是以积分或变分方式约束控制方程,因此在噪声和稀疏采样条件下更加稳健,并已在若干实验和生物场景中成功实现方程发现。然而,它们仍然依赖函数库设计和可观测性,同时还对测试函数选择、噪声建模和弱残差相关性建模敏感,这可能限制其在复杂生物数据中的可靠性。
总体而言,显式微分驱动的回归发现方法在概念验证和低维系统中表现突出,但在真实生物场景中,即使是现代弱形式变体,也会受到有限观测、模型错设和数据异质性的挑战。将回归与机制先验或降维方法结合的混合方法,可能是克服这些局限的重要方向。
其他回归方法
除了多项式展开和稀疏回归,其他机器学习技术也被用于动力系统研究。核方法,例如支持向量机或NARMAX,可以通过核函数将数据映射到非线性特征空间,从而建模非线性状态转移。这些方法已用于动力学状态预测和分类,也用于推断基因调控网络以及代谢网络、蛋白质相互作用网络等更广泛的生物网络。
高斯过程回归则为从数据学习动力系统提供了灵活的非参数贝叶斯框架。它能够给出带不确定性量化的概率预测,并已用于系统生物学中捕捉基因网络和信号通路的噪声动力学。不过,支持向量机和高斯过程回归通常更像黑箱预测器,而不是可解释动力学模型,并且在极高维或长时间序列数据中扩展性较差,因此更适合低维或机制相对明确的生物系统。
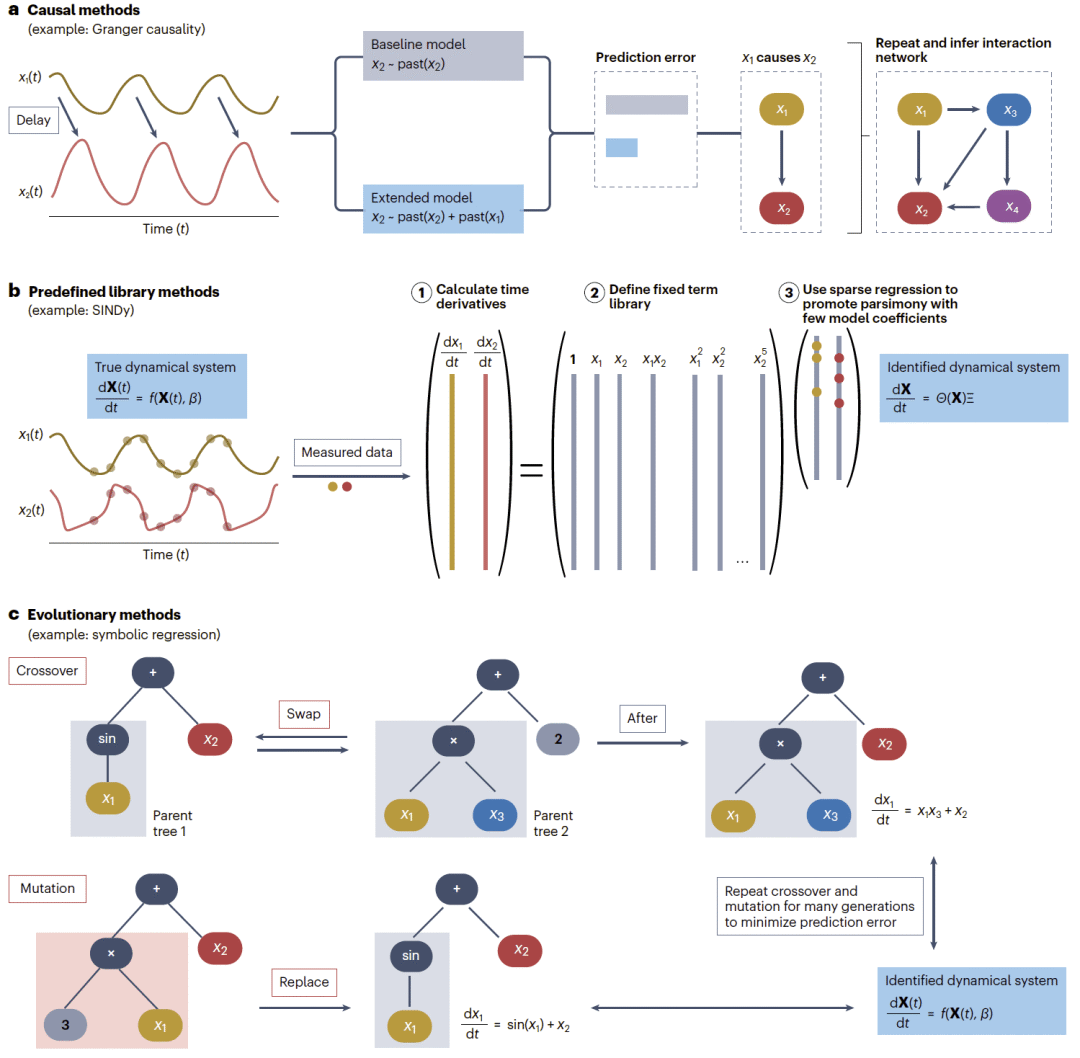
图2|用于动力系统识别的基于回归方法。
基于网络的方法
与依赖显式符号结构表示动力学的回归方法不同,人工神经网络是灵活的高维函数近似器,能够直接从数据中捕捉非线性动力学。它们的优势在于表达能力强,可以在不预设方程的情况下近似任意复杂映射,因而适合控制规则未知或只能部分观测的系统。然而,这种灵活性也有代价:神经网络通常是黑箱,需要大量数据,且解释性有限。回归方法产生显式方程,而神经网络则发现潜在表示,这些表示未必与可解释的可观测量对应。从Koopman理论看,神经网络可以被视为学习Koopman嵌入的有限维近似,但这种近似往往缺乏显式解释性。
研究人员重点讨论与动力系统最相关的三类神经网络架构:前馈神经网络、循环神经网络和自编码器。这些架构也构成了物理信息神经网络和生物信息神经网络等专门变体的基础,后者旨在平衡预测能力与解释性。
前馈神经网络
前馈神经网络是最简单的神经网络架构,用于将当前状态映射到下一个状态。网络通过学习从当前系统状态预测下一时刻状态,近似控制方程在一个时间步长内的流映射。
前馈神经网络已被用于振荡系统和混沌系统,也被用于生物学中的脑发育时间建模、基因调控网络和细胞间信号传导。从Koopman视角看,前馈神经网络可以被看作以黑箱方式隐式发现非线性可观测量,类似SINDy中的多项式库,但不是人为指定。一个常见局限是长轨迹预测中的误差累积,这可能使预测逐渐漂移到物理上不合理的区域,例如产生负浓度。物理信息神经网络和生物信息神经网络通过将机制约束直接嵌入损失函数来缓解这一问题,例如质量守恒或米氏动力学,从而提高物理合理性和训练域外泛化能力。
前馈神经网络也不仅可用于预测,还可用于发现相空间的静态几何特征。CLINE方法训练前馈神经网络来近似变量之间的反向关系,并通过评估导数为零的条件直接从数据中恢复零增长线结构,而不需要机制模型。由于零增长线定义了系统相图中的交点和稳定性结构,这种方法能够提供超越时间序列预测的机制洞察。
循环神经网络
循环神经网络通过引入环路获得记忆,因此天然适合处理时间序列和顺序数据。每个隐藏状态不仅取决于当前输入,也取决于前一个隐藏状态。这种递归结构使循环神经网络能够捕捉跨时间依赖,而前馈神经网络则独立处理每个状态。
循环神经网络已被用于混沌吸引子、振荡系统和系统识别任务。在生物学中,它们被用于建模基因调控动力学、细胞周期进程、湖泊温度剖面、微生物组动力学和微生物群体行为。
水库计算是循环神经网络的一个常用特例,其中循环水库保持固定,仅训练输出权重。由于只优化输出层,水库计算降低了计算成本并避免不稳定,同时仍保留较强表达能力。从Koopman角度看,循环神经网络和水库计算都可被理解为编码时间延迟坐标的灵活方式,类似于回归方法中的显式滞后项。
自编码器
自编码器通过将输入映射到低维潜变量,并从该潜变量重构原始状态,从而学习压缩表示。若使用线性激活,这一过程等价于主成分分析;若使用非线性激活,则可以发现更丰富的特征空间。
在动力系统中,自编码器提供了一种将高维动力学压缩为低维潜在坐标的方法,也可用于近似Koopman特征函数。其生物学应用包括细胞命运动力学、微生物生长、分子模拟和高维组学数据分析。在这一背景下,标准自编码器主要作为表示学习工具,用于定义低维状态空间,使系统动力学能够在其中进一步分析或建模。
变分自编码器是自编码器的重要扩展,它为潜在空间引入概率结构,并允许对数据分布进行生成式建模。其正则化机制鼓励潜在分布接近简单先验,抑制碎片化或高度弯曲的嵌入,并促进平滑、近似线性的潜在动力学。这种偏向低复杂度演化的性质有助于发现近似Koopman不变坐标,使非线性动力学在潜在空间中接近线性,因此特别适合与Koopman建模结合。与回归方法中的多项式展开类似,自编码器或变分自编码器发现的潜变量可被视为近似提升函数,只不过它们是直接从数据中学习得到,而不是预先指定。主要缺点在于对潜在维度敏感:维度过低会遗漏关键模态,维度过高则容易过拟合。
局限与展望
神经网络方法仍面临三类主要挑战。第一是解释性不足。高维嵌入通常很难对应明确的生物机制意义,与回归得到的符号模型形成鲜明对比。第二是依赖架构。模型表现高度依赖网络结构和超参数,常需要昂贵的反复试验。第三是泛化能力有限。尤其面对噪声较大或采样不足的生物数据时,训练域外泛化往往较弱。
Oregonator模型的比较结果突出了这些挑战。前馈神经网络在理想条件下可以复现训练窗口内的定性振荡行为,但面对不同初始条件时容易泛化失败;更换激活函数或缩小网络规模,也可能使网络无法捕捉振荡特征。
未来的一个方向是使用具有领域知识约束的变体,例如物理信息神经网络、生物信息神经网络和受Koopman启发的变分自编码器。这些方法可以在灵活性和可解释性之间取得平衡,但需要指定适当先验知识。总体而言,回归方法和网络方法代表了一个谱系的两端:前者强调符号可解释性,后者强调灵活近似。两者都可以统一在Koopman框架下,但在生物应用中具有不同优势和局限。
其他神经方法
除上述架构外,还有几类神经方法对动力系统建模越来越重要。神经常微分方程将离散映射替换为连续时间形式,用神经网络参数化导数,从而实现连续时间模型的端到端训练。图神经网络将标准网络扩展到关系数据,适合具有显式相互作用图的系统,例如基因调控网络或蛋白质相互作用网络。Transformer架构也正在被用于动力学预测,其自注意力机制能够比循环单元更有效地捕捉长程时间依赖。总体而言,这些方法扩展了前馈网络、循环网络和自编码器之外的表示工具箱:神经常微分方程提供连续时间动力学,图神经网络编码关系结构,Transformer擅长捕捉长程依赖,各自针对复杂生物系统中标准架构难以处理的问题提供了解决方向。
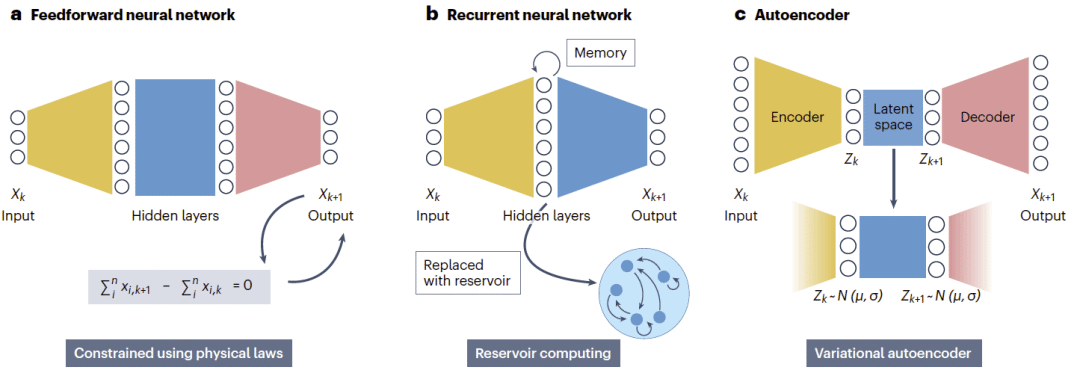
图3|用于动力系统的神经网络架构。
分解方法
在讨论神经网络如何通过高维嵌入近似Koopman算子之后,研究人员转向分解方法。这类方法能够从测量数据中更直接地近似Koopman算子。最常用的方法是动态模态分解,它最初用于从高维数据中提取时空相干结构,尤其是在流体动力学中。概念上,动态模态分解与适当正交分解有关,后者识别主导空间结构,而前者进一步编码这些结构的时间演化。因此,动态模态分解可以被解释为仅使用测量状态变量对Koopman算子的有限维近似。
动态模态分解
动态模态分解的核心思想是分析系统状态的连续快照之间如何相关。给定相邻时间点的系统快照,方法寻找一个线性算子,使其尽可能把前一组快照映射到后一组快照。这个线性算子因此提供了Koopman算子的有限维近似。
对于高维系统,直接计算完整线性算子通常不可行。因此,动态模态分解会将系统投影到由奇异值分解得到的低维空间中,使计算可行。保留多少模态需要谨慎选择:模态太少会丢失动力学信息,模态太多则会放大噪声。
精确动态模态分解的一个关键局限是依赖线性可观测量,而线性可观测量很少能够充分描述非线性动力学。扩展动态模态分解通过将数据提升到由非线性函数构成的高维特征空间中,再执行动态模态分解,从而缓解这一问题。这与SINDy等回归方法中使用候选基函数库的思想类似,并且这种联系在混合方法中被显式利用。
在生物场景中,动态模态分解和扩展动态模态分解已被用于健康和癫痫状态下的神经动力学、微生物生态系统、代谢动力学、血流和肿瘤生长等研究。这些应用表明,分解方法有潜力从复杂生物数据中揭示相干时空模式。
分解方法的局限
尽管分解方法具有吸引力,但在生物环境中仍面临两个主要挑战。第一,它需要足够丰富的动力学。由于动态模态分解和扩展动态模态分解通常用一个全局线性算子拟合整个数据集,数据必须包含多样的动力学状态,才能得到有意义的近似。当动力学主要由切换、噪声或短暂瞬态主导时,这些方法很难捕捉完整动力学谱,而这些情况在生物数据中很常见。虽然存在抗噪变体,它们仍然需要较高时间分辨率,而这在生物学中并不常见。
第二,它们在解释性方面依赖先验知识。扩展动态模态分解的成功取决于是否选择了合适的非线性提升函数,这一挑战与SINDy等回归方法类似。若选择不当,得到的模态可能缺乏机制意义,从而削弱生物解释性,并限制其与底层Koopman结构的联系。Oregonator模型中的示例进一步说明了这一点:当提升函数不足、数据质量低或测量噪声增加时,动态模态分解的表现会显著下降。
其他分解方法
虽然该领域主要由动态模态分解和扩展动态模态分解主导,其他分解技术也与动力系统相关。适当正交分解可以提供低秩空间模态,但忽略时间演化,因此常作为降维预处理工具。非负矩阵分解可从基因表达动力学和成像数据中提取可解释的加性组分。张量分解方法,如Tucker分解和相关的CANDECOMP/PARAFAC方法,则将这些思想扩展到多路数据,例如空间、时间和条件共同组成的数据结构,并越来越多地用于多组学轨迹分析。这些方法以不同方式在解释性、时间结构和计算成本之间进行取舍,因而可作为动态模态分解的补充。
与动态模态分解密切相关的还有基于延迟嵌入的分解技术。这些方法不仅试图重构系统动力学行为,还希望为恢复出的维度提供有意义解释。根据Takens嵌入定理,一个单一可观测量的时间延迟嵌入在一般情况下可以重构吸引子的拓扑结构,也就是说,即使某些维度不可观测,也可以通过延迟坐标展开隐藏维度,并定义基于测量的唯一坐标系统。这类方法在扩展动态模态分解无法找到有限维Koopman不变子空间时尤其有用。原则上,它们允许从部分观测中重构缺失状态变量,而这种情况在生物系统中非常常见。不过,从实验数据中识别合适嵌入并不容易。已有研究表明,延迟嵌入可从部分观测中重构缺失状态变量,使SINDy能够从不完整测量中识别肿瘤生长动力学的控制方程。
展望
推动数据驱动建模的实验前沿
建模中最紧迫的限制并不仅来自方法本身,也来自实验条件,因为生物系统通常高维、有噪声且只能部分观测。显微成像技术的进步正在提供更高的时空分辨率,使细胞和亚细胞过程能够被更细致地观察。动态生化测量的特异性和灵敏度也在荧光报告系统和基于FRET的探针等传感器帮助下不断提高。类似地,多组学创新有望提供更丰富的分子时间序列,尽管当前技术仍然较慢且往往具有破坏性,这使时间重构变得复杂。
一些分析流程已经开始通过将时间分辨测量和多组学测量整合进统一建模框架来应对这一挑战,为高维动态推断提供实际工具。另一个新兴方向是使用合成替代系统,例如能够复现部分生物动力学、同时提供完整实验控制的最小人工细胞。这些实验进展将产生越来越复杂的数据集。数据驱动方法面临的挑战,是如何有效利用这些数据,同时处理高维信息并产生可解释洞察。
伪时间与轨迹推断
伪时间方法广泛用于单细胞生物学,它们基于转录组相似性从静态群体数据中推断时间排序,为细胞分配潜在时间坐标。RNA velocity和最优传输分析等扩展方法进一步加入方向性和粗略速率信息,尽管RNA velocity估计的准确性仍在持续改进中。这类方法已经成为发育背景下映射细胞状态、分支拓扑和罕见转变的重要工具,并已有许多可用软件。
这些方法本质上是拓扑性的:它们恢复细胞状态空间中轨迹的形状和分支结构,但不推断驱动转变的物理力或速率。因此,它们不属于本文所讨论的Koopman框架。由于伪时间并不等同于真实物理时间,导数定义不良,SINDy或动态模态分解等基于回归或分解的方法也不能直接应用。虽然一些偏差感知方法可以部分缓解预处理选择带来的敏感性,但当系统动力学具有振荡特征时,伪时间方法的表现尤其受限。尽管如此,伪时间仍然是本文所讨论方法的重要补充,因为它能够提示动态变化发生的位置,从而指导后续有针对性地采集真实时间序列数据,用于机制建模。
势能景观方法
势能景观方法为细胞状态空间提供了更明确的动力学刻画。与伪时间不同,这类方法植根于非平衡统计物理,并且本质上具有概率性,因此在概念上不同于本文关注的确定性Koopman框架。它们不是简单绘制轨迹拓扑,而是量化塑造细胞命运的物理力,直接从数据中恢复固定点、转变屏障和由通量驱动的动力学。
在非平衡系统中,驱动力可以分解为势能景观梯度和旋转通量项。后者对于细胞周期等振荡和循环过程至关重要,因为仅靠梯度景观不足以解释循环动力学。随后发展出的多种互补框架,从基于突变理论的几何方法,到基于Fokker–Planck方程的数据驱动路径积分形式,再到用于最小作用路径和关键转变热力学早期预警指标的分解方法,均扩展了这一思想。
与伪时间相比,景观方法能够恢复更丰富的动力学结构;但与回归或分解方法相比,它们在相互作用网络推断和预测方面仍较弱。神经流图等新兴方法提示,未来可能形成混合框架,将概率景观概念与回归、网络和分解方法的机制可解释性连接起来。
结合优势的混合方法
新的方法越来越多地将回归与神经网络架构结合起来。这两类方法具有互补性:回归强调可解释性,但难以处理高维数据;神经网络灵活性强,却常常表现为黑箱。混合方法试图同时利用二者优势。
虽然也可以与分解方法结合,但显式整合分解方法的案例仍较少。更多时候,分解思想被嵌入自编码器等网络架构中,形成许多现代混合方法的基础。早期生物系统应用已经显示出较好前景。研究人员重点介绍了几种代表性方向。
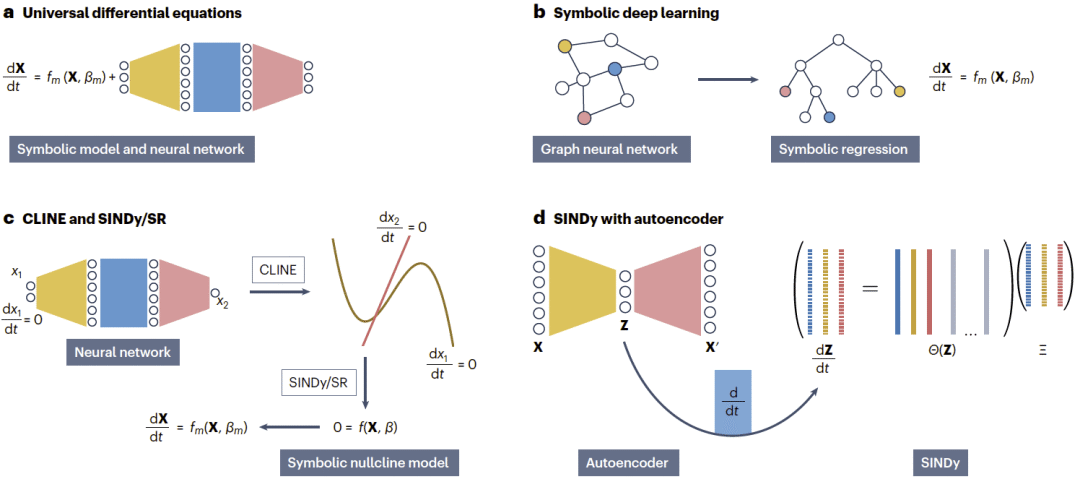
图4|整合基于回归和基于网络方法的混合方法示例。
通用微分方程直接将神经网络嵌入机制模型中。系统中已知的部分由经典微分方程描述,而未知或理解不充分的部分则由神经网络表示。这种方法允许端到端训练,同时保持时间一致性。对生物学而言,它非常有吸引力,因为许多系统只是部分被理解:已知反馈环可以显式建模,残余项则用于捕捉缺失生物学过程。其应用包括基因调控网络、流行病动力学和生态相互作用。
符号深度学习采取不同策略:它不是将神经网络嵌入方程,而是将训练好的网络转化为符号表达式。通常先由图神经网络学习相互作用规则,再使用符号回归提取可解释方程。这种方法结合了神经网络的表示能力和符号模型的解释性,并已用于生化振荡、群体动力学和药代动力学等问题。
CLINE-SINDy或CLINE-符号回归方法则利用CLINE无模型地发现相空间几何特征,如零增长线。一旦这些结构被识别,就可以通过SINDy或符号回归转化为显式方程。这种策略将任务从完整时间轨迹拟合转化为几何曲线拟合,从而提高鲁棒性。基于实验生化数据的结果表明,CLINE-符号回归能够识别强非线性函数,显示出基于零增长线的混合方法的潜力。
SINDy与自编码器的结合也具有重要意义。SINDy难以处理高维数据,而自编码器擅长将动力学压缩到低维空间。因此,将二者结合可以让自编码器发现降维坐标,再由SINDy在潜在空间中识别可解释的控制方程。这一平衡已被用于分析秀丽隐杆线虫运动等问题。此外,该框架还可以与发现缺失状态变量的方法结合,例如延迟嵌入技术,它可以从有限观测中重构高维系统。这样的整合使SINDy–自编码器框架特别适合测量稀疏且不完整的生物和生化系统。
生物学中数据驱动方法的未来目标
生物学中动力系统数据驱动方法的主要目标,是在底层生物过程背景下更深入理解动力系统。这种理解可以体现为预测系统行为、发现隐藏相互作用,或确定生物系统中的相关状态变量。本文介绍的大多数方法都能实现其中一个或多个目标。然而,在生物学中,尤其需要能够整体捕捉、描述并整合目标系统所有底层机制的工具。
研究人员认为,结合方法比较结果来看,这一任务最适合由微分方程形式的符号数学模型完成。这类模型能够显式描述相互作用、机制以及它们对系统状态的影响。若模型选择得当,它不仅可以预测系统行为,还可以探索“如果……会怎样”的情景。因此,一个好的模型甚至可以在实验进行之前,通过计算预测未知生物学。当然,这些预测最终必须经过实验验证。但这并不意味着模型本身是绝对正确的;所有模型都有局限,只是其中一些非常有用。事实上,数学建模已经多次在揭示生物机制中发挥关键作用,数学在生物学中的重要性也已被广泛认可。
微分方程模型适用于多种场景,例如基于常微分方程的描述可以捕捉状态空间中的有效调控和网络动力学,而空间扩展的反应–扩散偏微分方程则可描述形态发生、组织力学和细胞内模式形成。从时空成像数据中数据驱动发现偏微分方程已经取得重要进展,尤其是在可获得密集、高分辨率场数据的条件下。不过,从实验生物数据中学习偏微分方程仍是一个独立且困难的反问题,因为生物成像往往只提供有限、有噪声且部分观测的场,同时空间耦合复杂且异质。因此,本文聚焦于从时间分辨生物测量中推断有效动力学模型,而不是完整场偏微分方程识别。
方法学进展显示,这一愿景正在不断演化。神经流图等新方法将神经常微分方程扩展到更具表达力的时间序列表示,多组学分析流程则开始整合多组学动态数据。与此同时,Waddington表观遗传景观隐喻推动了从数据中直接重构发育景观的机器学习方法。这些努力表明,不同研究方向正在汇合到混合模型上,即同时连接机制可解释性和数据驱动灵活性的模型。
展望未来,数据驱动生物学不会由某一种单一方法主导,而会由混合方法推动。这些方法将结合符号方程的可解释性、神经网络的灵活性和概率框架的鲁棒性。它们或许永远无法给出绝对意义上“正确”的模型,但可以极其有用。通过从数据中提炼控制方程、发现隐藏状态变量,并将机制先验与机器学习结合,下一代模型不仅能够描述已知内容,还能预测未知现象,从而指导实验并塑造新的生物学理论。
整理 | DrugOne团队
参考资料
Prokop, B., Gelens, L. Data-driven discovery of dynamical models in biology. Nat Rev Phys (2026).
https://doi.org/10.1038/s42254-026-00955-4

本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号