Chem. Sci. | Pareto 是否是分子优化的正解?

Chem. Sci. | Pareto 是否是分子优化的正解?

MindDance
发布于 2026-06-29 14:17:53
发布于 2026-06-29 14:17:53
做药的人都知道一个朴素的难处:你想要的东西,往往互相打架。
一个理想的候选分子,既要和靶点结合得足够紧,又要溶得进血液;既要能穿过血脑屏障,又不能毒到正常细胞;既要结构新颖到能申请专利,又要简单到工厂愿意合成。每一项单独看都不难,难的是它们凑在一起——把结合力推高一点,溶解度可能就塌了;把分子做小一点好合成,活性又可能不够。
计算机辅助药物设计这些年最热闹的方向,是用深度生成模型去"凭空"造分子。但这条路有个绕不开的成本:要喂海量数据,要烧巨量算力。与此同时,一类更朴素的老办法——基于分子图的遗传算法——被开源出来之后,反而显出了它的好:不用训练,简单,稳,而且在不少基准上比深度模型更高效。
这篇发表在《Chemical Science》上的工作,做的就是把这条朴素路线往前推一步。作者 Jonas Verhellen 的思路很清楚:既然现实里的分子设计本来就是多个目标的权衡,那为什么还要把这些目标硬捏成一个分数去优化?不如直接去找那一整族"无法再两全其美"的解。
〔关键洞察〕 单目标优化是在问"哪个分子最好",多目标优化是在问"在所有无法同时改进的权衡点上,分别是哪些分子"。前者给你一个答案,后者给你一整条可供挑选的前沿。对一个还没想清楚自己要在结合力和毒性之间怎么取舍的药化学家来说,后者要有用得多。
下面我们把这套方法的来龙去脉讲清楚——它怎么定义"最好",怎么在矛盾中做选择,以及它跑出来的结果里,藏着一个相当反直觉的发现。
01 先把"最好"重新定义一遍:Pareto 前沿
传统做法是这样的:你有五个目标,那就给每个目标打个分,再用算术平均或者几何平均把它们揉成一个总分,然后优化这个总分。想让某个目标更重要?给它加个权重。
这套办法的问题在于,权重是你拍脑袋定的。可在很多真实项目里,你根本还不知道该怎么权衡——到底是宁可牺牲一点活性换更好的代谢稳定性,还是反过来?这个答案常常要等你把候选分子拿在手里、看到具体长什么样,才能下判断。
Pareto 优化绕开了这个困境。它不替你做权衡,而是把所有"不吃亏的权衡点"都摆给你看。
什么叫不吃亏的权衡点?一个分子如果存在另一个分子在每一项目标上都不比它差、且至少在一项上更好,那它就是被支配的,可以淘汰。反过来,找不到任何别的分子能在所有目标上全面碾压它——这样的分子就是 Pareto 最优。把所有 Pareto 最优解放在一起,它们在目标空间里连成一道边界,这道边界就叫 Pareto 前沿。
〔打个比方〕 想象你在买二手车,只关心两件事:便宜和省油。有的车又便宜又费油,有的车又贵又省油。Pareto 前沿就是那条"已经没法再占便宜"的边界——线上的每辆车,你想让它更便宜,就得忍受它更费油;想让它更省油,就得多掏钱。线下方的车则纯属冤大头,总能找到一辆在两方面都不输它的。Pareto 优化交给你的,是整条边界,让你自己根据预算和通勤距离去挑。
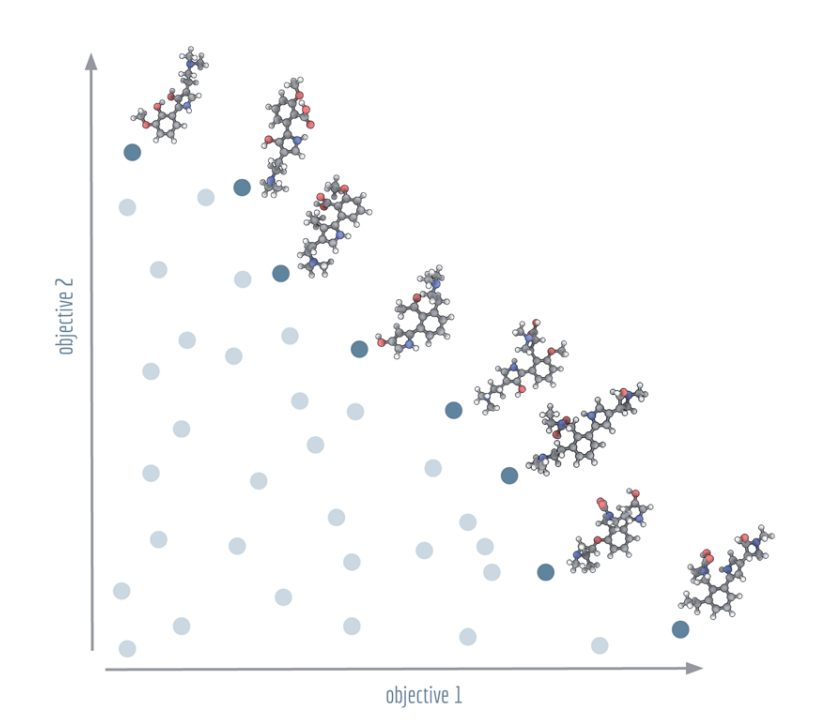
深蓝色点连成的 Pareto 前沿与浅蓝色的被支配解。这张图里的两个目标,是候选分子分别与麦角酸二乙酰胺、裸盖菇素的结构相似度——前沿上挂着的那些分子结构,正是算法在两个相互拉扯的目标之间找到的一系列权衡点。
深蓝色点连成的 Pareto 前沿与浅蓝色的被支配解。这张图里的两个目标,是候选分子分别与麦角酸二乙酰胺、裸盖菇素的结构相似度——前沿上挂着的那些分子结构,正是算法在两个相互拉扯的目标之间找到的一系列权衡点。
这就是非支配排序遗传算法(NSGA)要解决的事。它有两代主力:NSGA-II 和 NSGA-III。本文的工作,是把这两代算法都嫁接到一个成熟的、基于分子图的遗传算法框架上,做成开源实现,再放到一组真实的药物设计基准上去较量。
02 遗传算法:让分子像生物一样进化
在讲 NSGA 的精妙之处前,得先交代它的底座——遗传算法本身。
它的灵感直接来自自然选择。算法维护一群"候选解",在分子设计里就是一群分子,称为种群。每一轮迭代(叫一代)里,算法用两种操作生成新分子:mutation(突变,对单个分子做局部改动)和 crossover(交叉,把两个分子的片段拼到一起)。生成一批新分子后,按表现好坏做选择,把种群砍回原来的大小。一代代下去,选择压力会把整个种群推向更优的地方。
分子在这里用分子图来表示。一个关键的工程约束是:只保留那些能正确地在 SMILES 字符串和分子图之间来回翻译的结构——这相当于一道现实性闸门,把化学上根本不可能存在的图直接挡在门外。初始种群通常从 ZINC、ChEMBL 这类公开数据库里随机取。
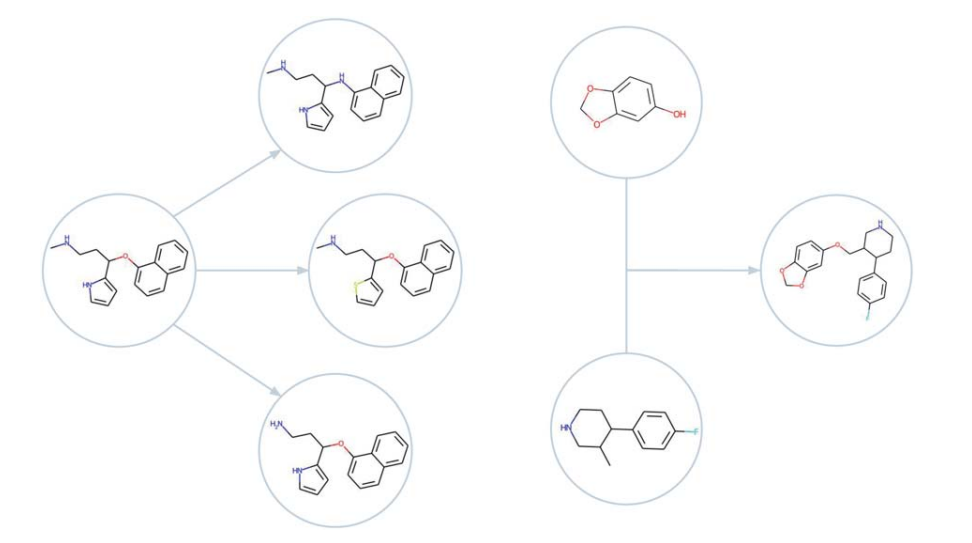
突变(左)与交叉(右)在分子图上的示例。值得注意的是,对结构做很小的改动,就足以推动优化向前走,哪怕面对的是很苛刻的目标。
突变(左)与交叉(右)在分子图上的示例。值得注意的是,对结构做很小的改动,就足以推动优化向前走,哪怕面对的是很苛刻的目标。
〔难在哪〕 遗传算法有个老毛病:进化停滞。当种群陷进某个表现平平的低谷,或者卡在局部最优里,它就可能原地打转出不来。单目标优化领域为此专门发展了一类"质量-多样性"算法——比如本文用作基线的 GB-EPI,它把种群按理化性质切成一个个小生境,每个生境只留最好的那个,强行维持多样性,逼着算法去探索更广的化学空间。
这里有个伏笔值得先记下:在单目标优化里,多样性是一种需要费力去维持的稀缺资源。而到了多目标优化,情况会反过来——这正是后面那个反直觉发现的根源。
03 当一代解太多,怎么取舍:分层与"分裂前沿"
NSGA 的核心动作,是不再用一个分数给分子排队,而是把整个种群按 Pareto 支配关系分层。
第一层(最重要的一层),是当前种群里完全不被任何分子支配的那些——它们就是当下的 Pareto 前沿。第二层,是只被第一层支配的那些。第三层,是去掉前两层后剩下的里头不被支配的那些。以此类推,整个种群被剥成一层层的"前沿"。
选择的时候,算法从第一层开始,整层整层地把分子接纳进下一代种群,直到快装满为止。
问题出在最后那一层。它往往装不下——这一层里的分子数量,超过了种群还剩的空位。这一层有个专门的名字叫 splitting front(分裂前沿)。麻烦在于,这一层里的分子彼此之间在 Pareto 支配的意义上是平起平坐的,没有谁明显比谁好。那到底留谁、弃谁?
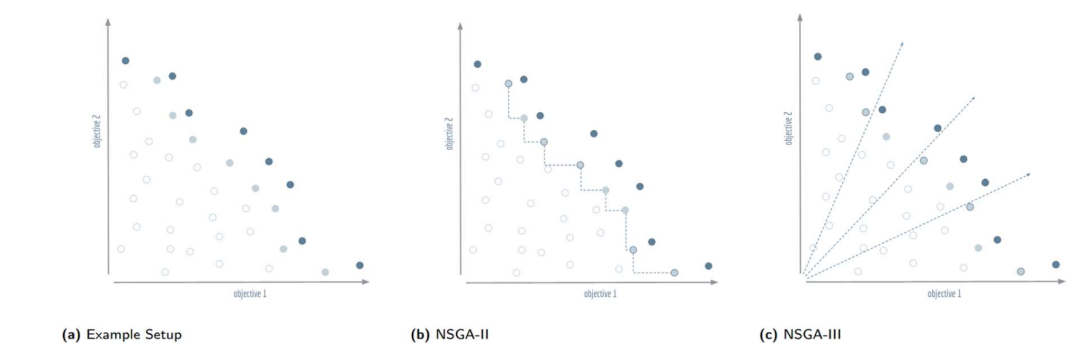
分裂前沿的处理示意。(a) 深蓝是当前 Pareto 前沿,浅蓝是这次需要做取舍的分裂前沿,白色是其余被淘汰的解,假设还需从浅蓝里挑出五个补满种群。(b) NSGA-II 的做法:算每个解到最近邻居的距离,优先留下周围最空旷的。(c) NSGA-III 的做法:算每个解到预设参考方向的垂直距离,每个方向各留最近的一个。
分裂前沿的处理示意。(a) 深蓝是当前 Pareto 前沿,浅蓝是这次需要做取舍的分裂前沿,白色是其余被淘汰的解,假设还需从浅蓝里挑出五个补满种群。(b) NSGA-II 的做法:算每个解到最近邻居的距离,优先留下周围最空旷的。(c) NSGA-III 的做法:算每个解到预设参考方向的垂直距离,每个方向各留最近的一个。
这道取舍题,就是 NSGA-II 和 NSGA-III 分道扬镳的地方。两者都靠"多样性"来做最后的筛选,但维持多样性的方式截然不同。
04 NSGA-II 的办法:谁周围越空旷,谁就留下
NSGA-II 用的是 crowding distance(拥挤距离)。
对分裂前沿里的每个分子,算法都算一个拥挤距离,衡量它被同伴包围得有多紧——具体用的是目标空间里的曼哈顿距离。拥挤距离越大,说明这个分子周围越空,越孤独。
逻辑很直白:周围越空旷的分子越该留。因为留下它,就能让最终的解在目标空间里铺得更开、更均匀,而不是挤成一团。算法把分裂前沿里的分子按拥挤距离从大到小排队,依次接纳,直到装满。两端最外侧的分子会被赋予无穷大的拥挤距离,确保它们每一代都被保住——不然前沿的边界会慢慢往里缩。
〔换个说法〕 NSGA-II 的取舍标准很本地化:它只看你在分裂前沿这一小撮分子里挤不挤,不关心整个种群的全局分布。这是个简单、便宜、好用的启发式。
05 NSGA-III 的办法:先画好辐条,每根辐条各留一个
NSGA-III 换了一套完全不同的思路:reference directions(参考方向)。
想象在目标空间里,从原点出发,预先撒下一把均匀分布的射线,像车轮的辐条一样朝各个方向延伸。算法把种群里每个分子,按它到最近那根辐条的垂直距离,归到对应的辐条上。做最后筛选时,算法优先照顾那些目前还没分到分子的辐条——哪根辐条空着,就从分裂前沿里挑一个离它最近的分子补上去。等所有空辐条都各自补了一个、种群还没满,剩下的名额就随机分配。
NSGA-II 和 NSGA-III 最本质的区别在这里:NSGA-II 的拥挤距离只在分裂前沿这一层内部计算,是个局部判断;而 NSGA-III 的参考方向考虑的是整个存活种群的全局分布。
〔一点判断〕 听上去 NSGA-III 更精细、更全局、更先进。事实上它确实是更新的一代,原本也是作为 NSGA-II 的改进版提出的。但论文很诚实地指出:后续大量数值实验表明,NSGA-III 并不在每个场景里都赢过 NSGA-II。这也是本文要把两者放在化学基准上正面比一比的原因——别想当然地以为新的就是好的。
06 把辐条摆均匀,本身是个难题
参考方向决定了 NSGA-III 最终解的分布,所以这些辐条撒得均不均匀,直接关系成败。
传统做法是 Das–Dennis 方法。但它有两个硬伤:一是因为它本质上是个组合构造,没法生成任意数量的方向——你想要多少根辐条,它给不了你随意指定的数目;二是它生成的方向大多擦着单纯形的边界走,而不是穿过内部,这会给分裂前沿的筛选引入偏差。
本文采用的是一种受物理学启发的新办法——能量法。它借用了势能的一个推广,叫 Riesz s-energy:把每两个参考点之间想象成带着同种电荷、互相排斥,距离越近排斥能越高。然后让所有点在单纯形上挪动,去最小化这股总排斥能。
〔Aha 时刻〕 这个类比很妙:一堆同性电荷在一个面上互相嫌弃,自然会把自己推到彼此最远、铺得最匀的位置上——就像几个人在电梯里会本能地站到对角。算法要的正是这种"自发铺匀"的效果。而且这套办法能生成任意数量、分布良好的方向,绕开了 Das–Dennis 的两个硬伤。论文里 s 取目标函数个数的平方根,沿用了原方法的建议。
07 一堆不起眼但要紧的工程细节
算法骨架之外,本文还借鉴 GB-EPI 加了一串小改进。这些东西单拎出来都不惊艳,但合在一起决定了实现好不好用:
突变和交叉是解耦的,分开施加而不是串成一串。原因是进化早期,交叉擅长大刀阔斧地探索化学空间;到了后期种群快收敛了,局部突变更适合精修。两者各管一段,分开用更顺。
突变操作被改造成做"位置类似物扫描"——这是把实验室里一种系统性改造分子的手法搬进了计算机,一次不只返回一个突变体,而是返回一个分子所有的位置类似物。这会带来额外计算量,于是又配了 memoisation(记忆化)来对冲:把算过的适应度记下来,碰到重复的分子直接查表,不重算。再加上目标函数评估的并行化,以及在分子进入评估之前先用 ADMET 结构过滤器把不合格的剔掉,整套实现的实际运行时间被压了下来。
〔具体一点〕 ADMET 过滤指的是吸收、分布、代谢、排泄、毒性这几方面的成药性筛查。在算法层面,它的作用是一道前置闸门:与其花算力去评估一个注定通不过成药性的分子,不如在它进评估之前就拦下来。这既省钱,也让最终吐出来的分子更靠谱。
08 基准怎么设计:让七道题逼近真实药物项目
要检验这套方法到底行不行,得有像样的考题。
本文从 BenevolentAI 的 GuacaMol 基准套件里,挑了五道针对真实药物的多目标优化任务——围绕五个已上市药物去微调结构和理化性质,这些任务都有三个或更多目标。在此之上,作者又自己设计了两道更贴近现实多靶点项目的题:
一道是多激酶抑制剂任务。目标是让分子同时抑制三个常与癌症相关的 DAP 激酶(DAPk1、DRP1、ZIPk),同时避开两个常见的脱靶离子通道(hERG、SCN2A)——hERG 尤其要躲,它跟心脏毒性密切相关。
另一道是抗精神病药任务。这反映了该领域近年的一个真实转向:从单一靶点转向同时结合多个受体。任务要分子同时结合两个血清素受体(5-HT2A、5-HT2B)和经典的多巴胺 DRD2 受体,同时躲开 hERG,还要满足辉瑞提出的中枢神经系统成药性要求。
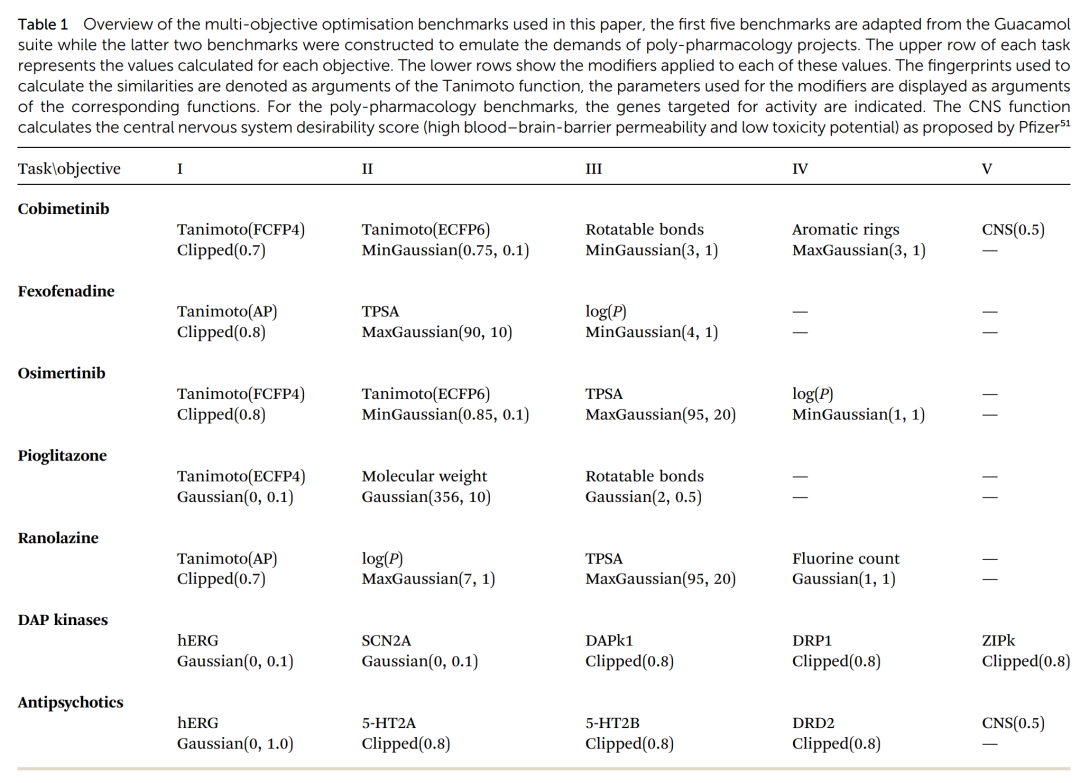
七道多目标优化基准的设计明细。每道题上行是要计算的各项目标(结构相似度、TPSA、logP、可旋转键数等),下行是把原始分数映射到 0 到 1 区间的修饰函数(Clipped 阈值型、Gaussian 靶向型及其 Min/Max 变体)。后两道多靶点题里,标注了要激活或要避开的具体基因靶点。
七道多目标优化基准的设计明细。每道题上行是要计算的各项目标(结构相似度、TPSA、logP、可旋转键数等),下行是把原始分数映射到 0 到 1 区间的修饰函数(Clipped 阈值型、Gaussian 靶向型及其 Min/Max 变体)。后两道多靶点题里,标注了要激活或要避开的具体基因靶点。
这里有一处细节值得读者留意:正文在介绍五个药物任务时点名了 perindopril(一种长效 ACE 抑制剂),但真正进入数据表格的第四道题是 pioglitazone。以实际跑出数据的表格为准来理解这套基准会更稳妥,这类正文与表格的出入,读原始论文时不妨多核一眼。
09 两把新尺子:超体积,和反直觉的相似度
要比较算法,光有题还不够,还得有量分数的尺子。本文引入了两把在化学优化里此前少见的度量。
第一把是 dominated hypervolume(支配超体积)。过去衡量多目标结果,习惯用几何平均。几何平均有个好处:它对任何单项的严重拉胯都很敏感,一项垮了总分就垮,这在很多场景下是合理的。但它本质上只盯着单个分子的综合分,看不出整条 Pareto 前沿的好坏。
支配超体积换了个视角:它把一组解在目标空间里支配的那片区域的"体积"量出来。以原点为参照下界,一组解的点越靠近理想前沿、铺得越开,它们罩住的体积就越大。这一个数,同时把"逼近程度"和"铺开程度"两件事都装进去了——正好是 Pareto 优化想要的。(顺带一提,目标数少于五个时这个体积能精确算,再多就得靠近似算法了。)
第二把尺子是用扩展指纹算的内部相似度,用来回答一个很关键的问题:这些算法是不是靠"更高的化学多样性"才赢的?传统上比多样性要两两算遍所有分子对的相似度,很费劲;本文用的扩展相似度指标能一次性比较任意多个指纹,不必算完整的相似度矩阵,效率高得多,而且退化到两个分子时就等于经典的相似度。文中具体用的是扩展 Faith 相似度指数。
10 结果:赢了基线,却没靠多样性赢
实验设置是每道题、每个算法各跑 20 次,每次 150 代,取均值和标准差。基线是 GB-EPI,它直接优化几何平均,代表单目标这一派的最强选手。
先看支配超体积,也就是 Pareto 前沿的整体质量。结果干净利落:在大多数任务上,NSGA-II 和 NSGA-III 都明显甩开了 GB-EPI 基线。几个有代表性的数字——
cobimetinib 任务:GB-EPI 是 0.77,NSGA-II 跳到 0.94,NSGA-III 是 0.92。ranolazine 任务:GB-EPI 只有 0.46,NSGA-II 到 0.68。这个差距是符合预期的:NSGA 这两位本就是冲着优化 Pareto 前沿去的,而 GB-EPI 优化的是几何平均,被一把专门量前沿质量的尺子比下去,理所当然。
但论文没有粉饰例外。在抗精神病药这道题上,NSGA-III 反而吃了亏(超体积 0.05,比 GB-EPI 的 0.09 还低)。原因是这道题的三个受体靶点彼此太像,而 NSGA-III 那套刚性的参考方向在这种情形下不灵光——预设的辐条方向,碰上目标之间高度相关的局面,反而成了束缚。
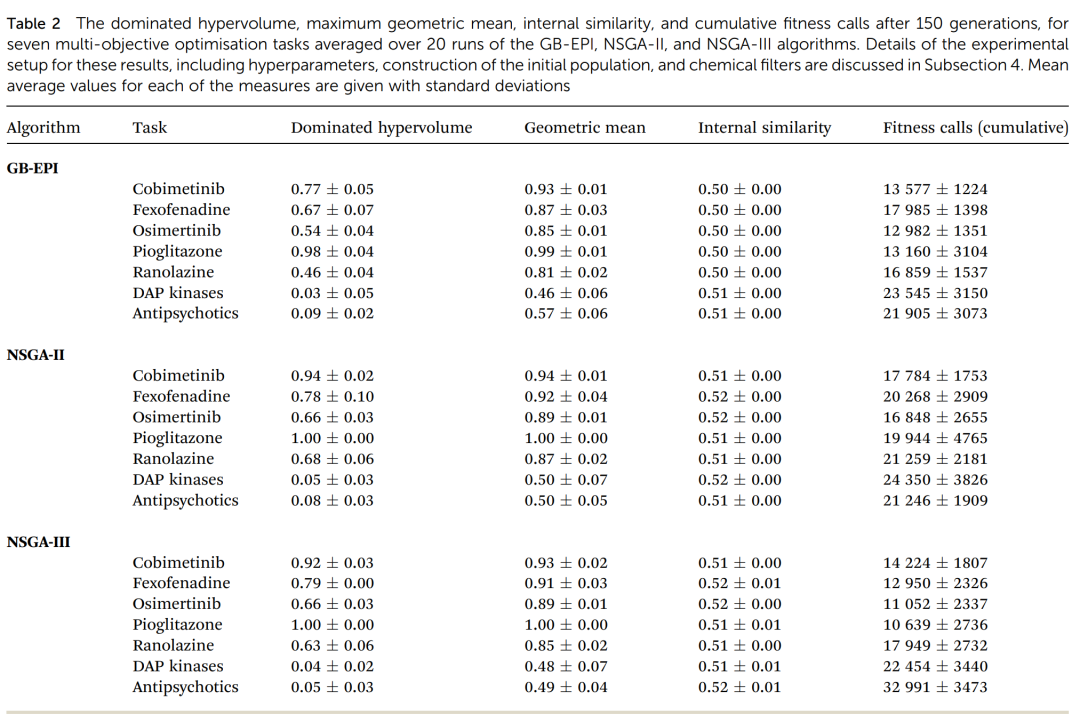
七道任务下 GB-EPI、NSGA-II、NSGA-III 三个算法的支配超体积、几何平均、内部相似度与累计适应度调用次数(20 次运行的均值±标准差,150 代)。这张表是全文的数据核心,三组数字横向对照,胜负、例外与效率差异一目了然。
七道任务下 GB-EPI、NSGA-II、NSGA-III 三个算法的支配超体积、几何平均、内部相似度与累计适应度调用次数(20 次运行的均值±标准差,150 代)。这张表是全文的数据核心,三组数字横向对照,胜负、例外与效率差异一目了然。
再看几何平均,趋势和超体积大体一致,但有个细节:各算法的几何平均最大值彼此挨得很近,在 cobimetinib 任务后期,GB-EPI 的 95% 置信区间和两个 NSGA 是重叠的。论文坦白指出,如果你的目的就是把几何平均推到全局最大、或者把某单个目标推到极致,那直接做单目标优化才对——Pareto 算法原则上也能摸到这些全局最优,但因为种群被摊开在整个目标空间里,效率会差不少。这是个诚实的边界声明:Pareto 优化不是万能药,它换来一整条前沿,代价是不再专注于某一个点。
〔边界〕 用一个聚合函数优化,解会扎堆在目标空间的一小块,覆盖不了整条前沿;用 Pareto 优化,解铺得开,但代价是不再死磕单点极值。选哪个,取决于你到底想要一个答案还是一族答案。这不是谁更强,是两种工具各有其位。
效率方面,本文追踪了 150 代里累计的函数调用次数。结论是 NSGA-III 在效率上整体最优——在那些它和别人performance相当的任务上,它用的调用次数最少。但同样有例外:还是那道抗精神病药任务,NSGA-III 的调用次数反常地飙到三万以上,远高于其他任务。性能和效率的两处失常都落在同一道题上,指向同一个症结——靶点太相似,刚性参考方向水土不服。
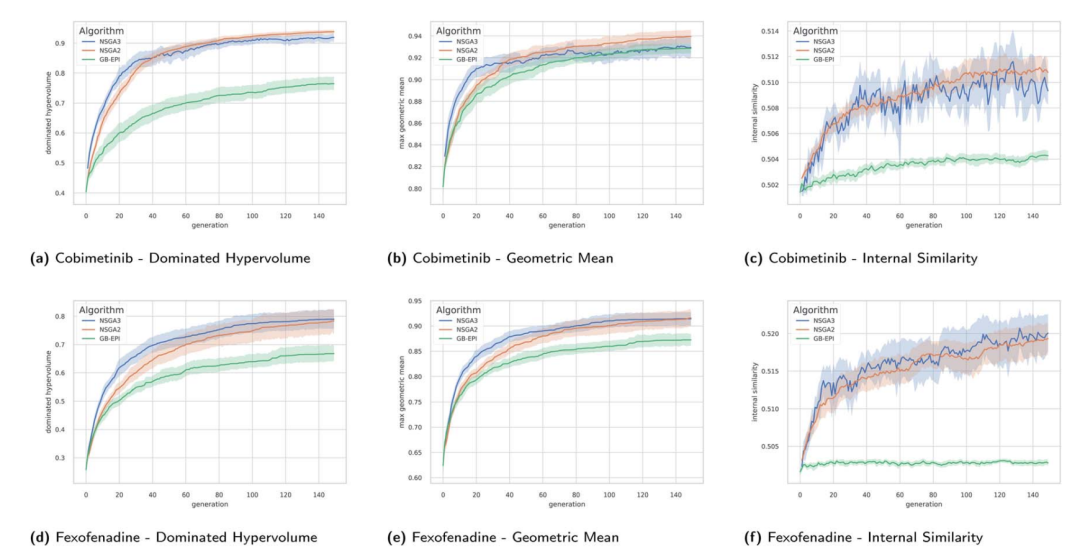
cobimetinib(上排 a–c)和 fexofenadine(下排 d–f)两道任务下,支配超体积、几何平均、内部相似度随代数演化的时间序列,带 95% 置信区间的方差带。橙色是 NSGA-II,蓝色是 NSGA-III,绿色是 GB-EPI。最值得盯着看的是第三列内部相似度:两个 NSGA 的曲线明显爬得更高、更快,而 GB-EPI 几乎贴在底下不动。
cobimetinib(上排 a–c)和 fexofenadine(下排 d–f)两道任务下,支配超体积、几何平均、内部相似度随代数演化的时间序列,带 95% 置信区间的方差带。橙色是 NSGA-II,蓝色是 NSGA-III,绿色是 GB-EPI。最值得盯着看的是第三列内部相似度:两个 NSGA 的曲线明显爬得更高、更快,而 GB-EPI 几乎贴在底下不动。
11 真正反直觉的,是那条相似度曲线
到这里,赢面已经讲清楚了。但这篇论文最有嚼头的发现,藏在内部相似度那一栏里。
按单目标优化的老经验,内部相似度低是好事——种群越多样,越不容易卡在局部最优,越能探索更广。GB-EPI 这类质量-多样性算法,整套设计就是为了维持低相似度、保住多样性。
可在多目标这边,结果反过来了:表现更好的 NSGA-II 和 NSGA-III,内部相似度反而更高,而且涨得更快。表里的数字差异不大(NSGA 多在 0.51 到 0.52,GB-EPI 多在 0.50),但方向极其一致——七道题里几乎条条如此,时间序列图上更是泾渭分明。
〔Aha 时刻〕 这个反转其实自洽得很。回想前面那个伏笔:单目标优化里,多样性是要费劲去挖的稀缺资源,因为你只奔一个目标,种群很容易扎堆;可在多目标优化里,多样性是天生就在场的——Pareto 前沿本身就是由各种权衡构成的,要满足互相打架的目标,解之间天然就得各不相同。 既然多样性已经被 Pareto 结构内建了,算法就不必再额外去强求它。这时候更高的内部相似度,反而说明种群成功地聚集在了高质量的前沿附近——大家都挤在好地方,而不是为了多样性而散在差地方。换句话说,在多目标的世界里,刻意维持多样性不再是美德,可能反倒是种拖累。
这是全文最值得带走的一句判断:单目标优化里被奉为圭臬的"多样性越高越好",到了多目标优化里需要被推翻重写。优化范式一换,连什么算"健康的种群"这种底层直觉都得跟着翻新。
12 把这套东西摆在更大的语境里
退一步看,这篇工作的分量不全在某个算法赢了几个点。
它真正的价值有三层。一是把成熟的多目标优化机器——NSGA-II、NSGA-III——完整、开源地搬进了分子设计,填上了一个空白:此前这两套算法用在分子设计上的,多是闭源的专有实现,社区拿不到趁手的开源版本。代码已公开在 GitHub 上,作者明确希望它能当作未来的基线和起点。
二是把支配超体积和扩展相似度这两把尺子引进了化学优化的评测体系。尤其是支配超体积,它比单看几何平均更能分辨一整条前沿的好坏,给这个领域的基准测试添了更有判别力的工具。
三是那个反直觉发现本身——它提醒所有做多目标分子优化的人,别把单目标时代的经验直接套过来。
作者也指了几条往下走的路:用上下文多臂老虎机或高斯过程,在分子进评估前先剪枝,进一步压低昂贵的函数调用;以及把这套算法接进自动驾驶实验室或主动学习的工作流里,让计算端的多目标优化和实验端的合成测试真正闭环起来。
〔一点判断〕 在深度生成模型吸走大部分注意力的当下,这篇工作有点逆流而上的意味:它说明一个不用训练、不烧数据的老派算法,配上对的问题框架(Pareto 而非单一聚合分数),照样能在真实的药物设计基准上打出漂亮的结果。它没有声称要取代深度模型,而是踏踏实实补上了一块开源的、可复现的、可当基线的基础设施——这种工作往往比又一个刷榜的新模型更经得起时间。
做药这件事,本质上从来不是把某一个指标推到极致,而是在一堆互相矛盾的诉求之间找一个能让所有人点头的平衡点。这篇论文做的,是把这种平衡的艺术,老老实实地交还给了算法该有的样子——不替你拍板权重,而是把所有不吃亏的选项,一并摆到你面前。
参考文献
Jonas Verhellen, "Graph-based molecular Pareto optimisation", Chemical Science, 2022, 13, 7526–7535.
https://doi.org/10.1039/D2SC00821A
Jonas-Verhellen/MolecularGraphPareto

本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号