不只是平均池化:EvoPool 如何把同源序列“压缩”进蛋白语言模型表示?

不只是平均池化:EvoPool 如何把同源序列“压缩”进蛋白语言模型表示?

Tom2Code
发布于 2026-07-01 20:38:52
发布于 2026-07-01 20:38:52
论文精读:EvoPool: Evolution-Guided Pooling of Protein Language Model Embeddings 作者:Navid NaderiAlizadeh, Rohit Singh 单位:Duke University 版本:bioRxiv preprint, posted February 4, 2026 doi:https://doi.org/10.64898/2026.02.02.703349

1. 这篇论文想解决什么问题?
蛋白质语言模型(Protein Language Models, PLMs)通常会把一条氨基酸序列编码成一串 residue-level embeddings:
其中, 是序列长度, 是每个残基 embedding 的维度。
但是很多下游任务并不是 residue-level 的,而是 protein-level 的,比如:
- 这个变体是否影响蛋白功能?
- 这个蛋白是否稳定?
- 这个蛋白是否具有某种结合能力?
- 这个突变相对于 wild type 的 fitness 会升高还是降低?
于是我们必须把 的 residue embedding 压缩成一个固定长度的 protein embedding:
最常用的方法是平均池化:
这个方法简单、稳定、便宜,但也有明显的问题:它默认每个残基同等重要。
然而真实蛋白功能往往由少数关键残基、保守 motif、结合界面、变构通路或结构核心决定。对长蛋白来说,平均池化尤其容易把关键残基信号“稀释”掉。
EvoPool 的核心问题可以概括为:
既然同源序列携带了进化约束,为什么 pooling 阶段仍然只看 query sequence 自己?
换句话说,作者不是重新训练一个 PLM,也不是把 MSA 塞进模型输入,而是提出:在 residue embeddings 已经算好的前提下,用 homolog embeddings 来指导 pooling。
2. 从平均池化到“进化引导池化”
假设我们有一条 query protein sequence:
以及它的一组同源序列:
注意这里的同源序列数量 可以变化,每条 homolog 的长度也可以不同。经过同一个 PLM 编码后:
EvoPool 想学习一个 pooling function:
这个函数必须满足三个不变性:
- 不依赖 query 序列长度 ;
- 不依赖 homolog 数量 ;
- 不依赖每条 homolog 的长度 。
这就很难了。因为输入不是一个固定矩阵,而是一个“变长序列 + 变数量 homolog 集合 + 每个 homolog 又变长”的复杂对象。
EvoPool 的答案是:把所有 residue embeddings 都看成高维空间中的点云,然后用 optimal transport 做几何池化。
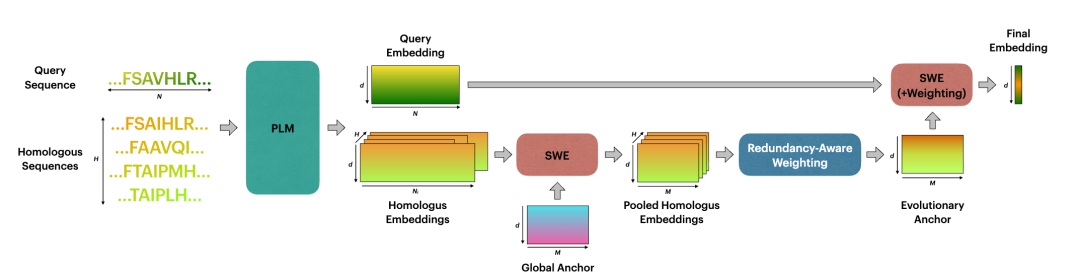
3. 核心数学工具:Sliced Wasserstein Embedding
EvoPool 建立在 Sliced Wasserstein Embedding(SWE)之上。
我们先忘掉蛋白,只看一个点集:
它可以代表一条蛋白的 residue embeddings。再给定一组 anchor points:
SWE 的思想是:高维 optimal transport 太贵,我们可以把高维点投影到一维,在一维上排序,然后计算“从 anchor 到 query 的位移”。
给定一个切片方向:
把 query 和 anchor 都投影到一维:
如果假设 query 和 anchor 数量相同,并且 anchor 投影已经升序排列:
再把 query 投影排序:
那么第 个切片上的 Monge coupling 可以写成:
直观理解: anchor 上第 个点,要移动多少,才能对齐 query 上排序后的第 个点?
多个切片组合起来,就得到:
如果再用一个权重向量:
就可以把它压缩成:
这就是 SWE pooling 的基本结构。
在 EvoPool 中,作者采用 axis-aligned slices:
也就是说,切片方向就是 embedding 的每个坐标轴。这样做的好处是:不需要学习切片方向,参数更少,更适合 task-agnostic 的自监督训练。
4. EvoPool 的两级结构:先造 evolutionary anchor,再池化 query
EvoPool 最漂亮的地方在于它使用了 two-level SWE。
4.1 第一级:把每条 homolog 压缩到统一尺寸
先定义一个可学习的 global anchor:
对第 条 homolog embedding:
使用 SWE 得到固定尺寸表示:
这一步解决了 homolog 长度不同的问题:不管 多长,每条 homolog 最后都变成 。
4.2 冗余感知加权:避免相似 homolog 重复投票
MSA 或 homolog 集合常常有冗余:某些分支被数据库过度采样。如果直接平均,密集谱系会被放大。
因此 EvoPool 先计算每个 homolog pooled embedding 的局部密度:
其中 是 cosine similarity, 控制密度估计的锐度。
然后用反密度权重:
并归一化:
于是 evolutionary anchor 定义为:
这个式子很关键。它不是简单求平均,而是让稀有、非冗余的 homolog 获得更高权重,让重复相似的 homolog 贡献更小。
可以把它理解成:
EvoPool 先把 homolog 集合压缩成一个“进化锚点”,这个锚点代表 query 蛋白所在蛋白家族的几何分布。
4.3 第二级:用 evolutionary anchor 池化 query
有了 evolutionary anchor 后,再对 query embedding 做一次 SWE:
最后通过权重向量:
得到最终 protein-level embedding:
整个 EvoPool 的可学习参数只有:
参数量为:
这也是它很实用的一点:相比重新训练 PLM,EvoPool 是一个轻量的 pooling module。
5. 自监督训练:让 embedding 既稳定,又不坍缩
EvoPool 不是用下游标签训练的,而是自监督训练。它的目标有两个:
- 同一个 query,换一批相似 homolog,得到的 embedding 应该一致;
- homolog 集合内部的表示不能全部塌缩成同一个点。
5.1 对比学习:同一个 query 的两种 homolog 子采样应该靠近
对每个 query ,随机采样两组 homolog:
得到两个表示:
它们应该靠近;而不同 query 的表示应该分开。于是使用类似 SimCLR 的 contrastive loss:
其中:
直观来说: 同一个蛋白的不同 homolog 子采样是正样本,不同蛋白之间是负样本。
5.2 Uniformity constraint:防止 homolog 表示内部坍缩
只做对比学习还不够。模型可能学到一种偷懒策略,让 homolog 相关表示变得过于集中。于是作者加入 uniformity constraint。
对某个 query 的两组 homolog 子采样,作者把每条 homolog 也当成 query,用另一组 homolog 作为上下文,得到一个集合:
然后定义 uniformity loss:
如果所有点都挤在一起, 很小,指数项大,loss 就偏高;如果点分散,loss 会降低。
论文把它写成约束:
这相当于告诉模型:
你可以让同一 query 的两个采样结果稳定,但不能把 homolog 空间压成一团。
5.3 Primal-dual optimization:约束学习的味道来了
最终优化问题是:
subject to:
作者用拉格朗日乘子:
构造 Lagrangian:
然后做 primal-dual 更新:
这个更新非常有意思: 如果某个 query 的 homolog embedding 发生坍缩,uniformity constraint 被违反,那么对应的 会变大,模型下一步就会更强地惩罚这种坍缩。
换句话说,dual multiplier 像一个自动调节的“警报器”:谁坍缩严重,谁就被重点约束。
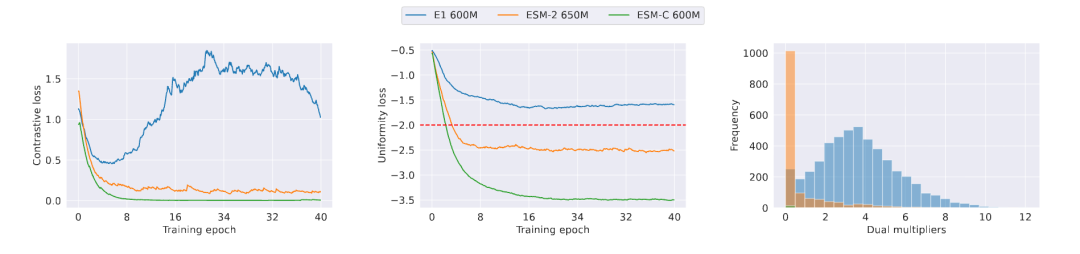
6. 实验设计:用 ProteinGym 做零样本变体效应预测
论文在 ProteinGym benchmark 上评估 EvoPool。这个 benchmark 包含大量深度突变扫描(DMS)实验,作者聚焦 substitution assays:
- 217 个 DMS substitution assays;
- 约 240 万个实验测量过的变体;
- 评价指标是预测分数与实验 fitness 的 Spearman correlation。
为了纯粹评估 pooling 的好坏,作者没有使用 likelihood-based variant scoring,而是用 wild type 和 variant 的 pooled embedding 的 cosine similarity 作为预测分数。
即对 wild type:
对 variant:
预测分数:
然后计算:
对比对象是最常见的 AvgPool。
实验使用了三个 PLM backbone:
- ESM-2 650M;
- ESM-C 600M;
- E1 600M。
训练阶段,作者从 OpenProteinSet 的 UniClust30 clusters 中随机选取 5000 个 MSA,homolog 数量从 8 到 24 中随机采样。主要超参数包括:
- ;
- ;
- contrastive temperature ;
- batch size = 8;
- training epochs = 40;
- primal learning rate = ;
- dual learning rate = 0.2;
- uniformity threshold 。
7. 结果:EvoPool 明显优于平均池化
从论文表格可以看到,EvoPool 在三个 PLM backbone 上都超过了 AvgPool。
以平均 Spearman correlation 为例:
PLM | AvgPool | EvoPool | 提升 |
|---|---|---|---|
E1 600M | 0.0857 | 0.2603 | +0.1746 |
ESM-2 650M | 0.2124 | 0.3423 | +0.1299 |
ESM-C 600M | 0.3760 | 0.4435 | +0.0675 |
这个结果说明一件事: 即使 PLM 已经在大规模序列上预训练过,pooling 阶段显式加入 homolog 信息仍然有价值。
更细看不同 function type,EvoPool 在 activity、binding、expression、organismal fitness、stability 等类别上都有提升。尤其是在 activity、expression 和 organismal fitness 上,提升更明显,这些任务往往更依赖进化约束和功能上下文。
论文还按 taxon 和 MSA depth 分层分析。结果显示,在 human、other eukaryote、prokaryote、virus 这些不同来源蛋白中,EvoPool 都有提升;在 low、medium、high MSA depth 下也都有提升。
这很关键,因为它说明 EvoPool 不是只在深 MSA 条件下有效。相反,作者强调它在 low/medium MSA depth 下依然能带来收益。
图 4 占位:结果表格可视化 建议把论文 Table 1 和 Table 2 重画成柱状图:横轴为 PLM backbone,纵轴为 Spearman correlation,对比 AvgPool 和 EvoPool。
<!-- 插入图4:EvoPool vs AvgPool across PLMs -->
8. 为什么长蛋白更受益?
论文还分析了 EvoPool 相对于 AvgPool 的提升和序列长度的关系。
结果发现:
- 对 E1 600M 和 ESM-2 650M,序列越长,EvoPool 的收益越明显;
- 对 ESM-C 600M,收益随长度变化更平稳,但整体仍有提升。
这个现象很符合直觉。
平均池化对长蛋白尤其不友好。假设真正有功能信息的残基只有 个,而蛋白长度是 。平均池化中关键残基信号大约被缩小为:
当 增大时,关键 motif、结合界面或变构残基的信号很容易被背景残基淹没。
EvoPool 的优势在于:它不是把所有残基平均看待,而是通过 evolutionary anchor 改变 query embedding 的几何对齐方式,让 homolog family 中保留下来的结构/功能约束参与 pooling。
可以粗略理解为:
而是:
它保留的是 query residue cloud 相对于 evolutionary anchor 的几何位移,而不是简单均值。
9. 同源信息放在输入端还不够,pooling 阶段仍然有价值
一个很自然的问题是: 如果 PLM 本身已经是 retrieval-augmented,比如 E1 可以在输入阶段使用 homolog,那 EvoPool 还有意义吗?
论文专门做了实验:在 E1 retrieval-augmented 模式下,比较:
- homolog 只用于输入检索,然后 AvgPool;
- homolog 用于输入检索,同时 pooling 阶段再用 EvoPool。
结果显示,在多数 DMS assays 上,第二种方式更好。
这说明:
输入阶段使用 homolog 和 pooling 阶段使用 homolog,并不是重复劳动,而是互补的。
输入端 homology 主要影响 residue embeddings 的生成;pooling 端 homology 则影响 residue embeddings 如何被聚合成 protein-level representation。
这也是 EvoPool 的核心位置:它不是替代 PLM,而是站在 PLM 之后,专门解决“如何把 residue-level 信息聚合成 protein-level 信息”的问题。
10. 我认为这篇文章真正的贡献是什么?
我觉得 EvoPool 的贡献可以分成三层。
第一层:把 pooling 从“算均值”变成“分布对齐”
平均池化只看一阶均值:
而 SWE 把 residue embeddings 看成一个 empirical distribution:
再通过 anchor 计算分布之间的几何位移。这比均值更丰富,因为它保留了 residue embedding cloud 的排序、形状和几何偏移信息。
第二层:把 homolog 信息放进 pooling,而不是只放进输入
已有 homology-aware PLM 多关注输入端,例如 MSA Transformer、PoET、E1 等。EvoPool 则关注一个不同问题:
给定 PLM 已经生成的 residue embeddings,如何在 pooling 阶段显式使用 homologs?
这个问题很重要,因为很多 downstream pipeline 已经有现成 PLM embedding,不一定能重新训练或改造模型输入。
第三层:用自监督 + 约束优化训练 task-agnostic pooling
EvoPool 没有依赖下游标签,而是通过:
保证同一 query 在不同 homolog 子采样下稳定;通过:
避免 homolog 表示坍缩;再通过 primal-dual optimization 动态调节约束强度。
这让它具备一定通用性:训练好后可以用于 zero-shot variant effect prediction。
11. 这篇文章也有什么局限?
我认为至少有四点值得注意。
第一,EvoPool 仍然需要 homologous sequences。虽然它不要求固定长度或固定数量的 homolog,但推理时仍然要能拿到 homolog 集合。
第二,论文主要评估 substitution DMS assays,对 indel、结构大扰动、多突变组合、蛋白-蛋白互作等任务还需要进一步验证。
第三,作者采用 axis-aligned slices:
这让模型参数更少、更稳,但也可能限制了表达能力。未来可以考虑 supervised fine-tuning 或 task-adaptive slicing directions。
第四,EvoPool 的核心计算依赖 homolog embeddings。如果 homolog 数量很大,虽然比 MSA Transformer 这类模型轻,但仍然存在检索和编码成本。
12. 对我们做蛋白 embedding 的启发
如果我们想设计一个新的 protein embedding 方法,EvoPool 给了一个很重要的启发:
创新不一定在 PLM 主干,也可以在 pooling 阶段。
尤其是当我们已经有 ESM、ProtT5、ESM-C、E1 这类强 backbone 时,重新训练一个大模型成本很高;但设计一个更聪明的 pooling module,成本低很多,也更容易和现有模型兼容。
基于 EvoPool,可以继续发展几个方向:
- 结构引导 EvoPool 用结构邻接图或接触图构造 anchor,而不仅仅是 homolog embeddings。
- 小样本任务自适应 EvoPool 在 self-supervised EvoPool 基础上,用少量下游标签微调 、 或 slicing directions。
- MSA-scarce EvoPool 当 homolog 很少时,用 sequence-derived pseudo-homologs、protein language model augmentation 或变体生成模型补充 family context。
- 多视图 anchor 同时构造 evolutionary anchor、structural anchor、thermodynamic anchor,再用 gating mechanism 融合。
- 可解释 residue transport 分析哪些 residues 在 Wasserstein displacement 中贡献最大,可能用于定位功能位点、保守 motif 或突变敏感区域。
13. 一句话总结
EvoPool 的核心不是“又提出一个 pooling trick”,而是提出了一种新的视角:
蛋白表示不应该只来自单条序列的 residue embeddings,而应该来自 query protein 相对于其 evolutionary family 的几何位移。
平均池化问的是:
这条蛋白的平均长什么样?
EvoPool 问的是:
这条蛋白的分布,相对于它的同源家族,发生了怎样的?
这就是它比普通 pooling 更有生物学意味的地方。
完~
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-06-30,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号