GeneKI:原文说“吊打”scTenifoldKnk|仅需WT scRNA数据直接进行目标基因虚拟敲除

GeneKI:原文说“吊打”scTenifoldKnk|仅需WT scRNA数据直接进行目标基因虚拟敲除

KS科研分享与服务-TS的美梦
发布于 2026-07-06 19:07:10
发布于 2026-07-06 19:07:10
1、GeneKI简介
GenKI是一款旨在仅利用野生型(WT)样本的单细胞RNA测序(scRNA-seq)数据进行虚拟敲除的工具,在没有KO样本的情况下预测基因功能,这与scTenifoldKnk(还来得及!虚拟敲除(1)-scTenifoldKnk包scRNA数据单基因虚拟敲除【详细解释】)是类似的,目的很明确简单,不同的是scTenifoldKnk是基于R语言的工具,GeneKI是基于python的工具。GeneKI工具2023年发表在Nucleic Acids Res杂志。按照原文说法及结论,GeneKI是吊打scTenifoldKnk的同类工具。
GenKI的设计目标是在完全不使用任何真实KO样本信息的前提下,以无监督方式捕捉由KO扰动引起的基因调控变化模式,为基因功能研究提供一个可扩展的框架。为实现这一目标,GenKI采用variational graph autoencoder(VGAE)模型,从输入的WT scRNA-seq数据和衍生的单细胞基因调控网络(scGRN)中学习基因的潜在表征以及基因间的相互作用。随后,通过计算方式从scGRN中移除待敲除基因(即拟进行功能研究的基因)的所有连边,从而生成虚拟KO数据。
论文原文(引用):Yang Y, Li G, Zhong Y, Xu Q, Chen BJ, Lin YT, Chapkin RS, Cai JJ. Gene knockout inference with variational graph autoencoder learning single-cell gene regulatory networks. Nucleic Acids Res. 2023 Jul 21;51(13):6578-6592. doi: 10.1093/nar/gkad450. PMID: 37246643; PMCID: PMC10359630.
GeneKI官网:https://github.com/yjgeno/GenKI/tree/master
GeneKI主要运行步骤及原理简析: 1、构建野生型单细胞基因调控网络(scGRN);输入野生型(WT)样本的单细胞基因表达矩阵,使用主成分回归计算基因间的相互作用强度,生成一个邻接矩阵网络。为了降低噪声,默认保留权重前15%的边,将其转化为布尔矩阵。 2、训练VGAE模型;利用基因表达矩阵以及构建后的网络,使用两层图卷积网络(GCN)作为编码器,学习每个基因在低维潜在空间中的表示,到基因的潜在分布参数,包括均值和方差。 3、模型参数迁移;冻结已经训练好的VGAE模型的权重,这个模型现在已经“理解”了野生型状态下的基因调控模式,将作为后续对比的基准。 4、构建虚拟敲除(KO)数据;确定要敲除的目标基因,在野生型网络的副本中,将所有与该目标基因相连的边全部删除(即权重设为0),生成虚拟敲除网络。 5、推断虚拟状态的特征表示;将基因表达矩阵和KO网络输入训练好的VGAE模型,得到在敲除背景下,每个基因新的潜在分布参数。 6、计算KL散度;对每个基因,计算其在WT状态和虚拟KO状态下潜在分布之间的Kullback–Leibler(KL)散度。KL散度值越高,说明该基因受敲除事件的影响越大。 7、鉴定对KO有响应的基因,用于功能注释和分析;为了提高稳定性,GenKI使用了基 Bagging(套袋法)的策略。通过多次排列(Permutation)细胞顺序并重复计算,最终筛选出在95%以上的次数中均进入前5%排名的基因。这些基因就是受虚拟敲除/扰动的基因,可以进行功能探究。
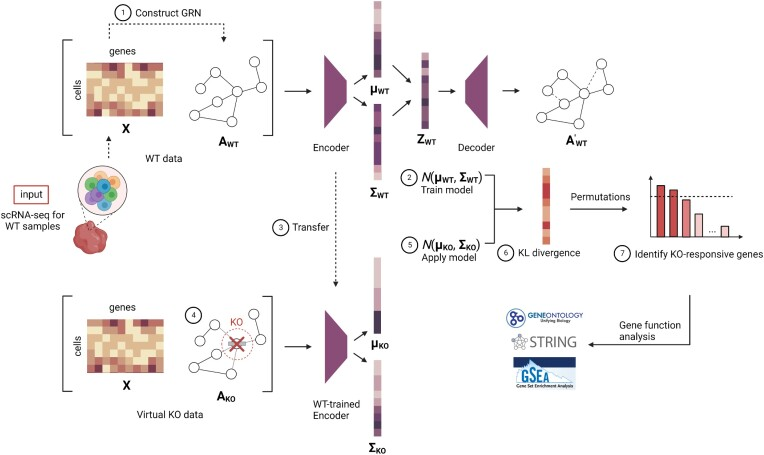
image-2.png
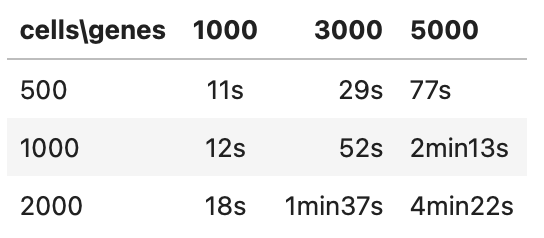
关于运行时间:
2、GeneKI及依赖包安装
#创建环境
#安装GenKI
#GenKI requires Python ≥ 3.10.
conda create -n geneki python=3.10
conda activate geneki
#安装
pip install scanpy -i https://pypi.tuna.tsinghua.edu.cn/simple
pip install GenKI -i https://pypi.tuna.tsinghua.edu.cn/simpleimport os
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
import scanpy as scimport GenKI as gk
from GenKI.preprocessing import build_adata
from GenKI.dataLoader import DataLoader
from GenKI.train import VGAE_trainer
from GenKI import utilsmicroglial_seurat_WT.h5ad是GeneKI包作者提供的演示数据(数据下载链接:https://drive.google.com/file/d/1tG9bUGCsWqhg0hJ94lDLtLl8WLl0hDks/view?usp=sharing),因为GeneKI是python工具,需要的输入数据是anndata,所以单细胞表达数据是h5ad形式,如果您的数据是seurat格式,可以进行转化。
#读入数据
adata = build_adata("./microglial_seurat_WT.h5ad")
adata = adata[:, :].copy()
adataadata.obs['celltype'] = 'microglial'#后续需要使用多线程,需要先安装:
pip install "GenKI[ray]"# load data — build the GRN and prepare WT / virtual-KO graph data
data_wrapper = DataLoader(
adata, # AnnData object,前面处理好的表达矩阵anndata
target_gene=["TREM2"], # KO gene name(s),需要敲除的任意基因名称
target_cell=None, # obs label for cell-type subset; None = all cells
#也即是说,你的anndata是所有celltype的时候,可以设置需要分析的celltype
#无需提取数据,这里因为是提取的数据,都是一种celltype,所以设置None
obs_label="ident", # adata.obs column for cell-type labels,细胞注释列
GRN_file_dir="GRNs", # folder for cached GRN .npz,储存GRN .npz的文件夹名称
rebuild_GRN=True, # build GRN if not cached
n_cpus=8, # Ray sharding; needs `pip install "GenKI[ray]"`
)
data_wt = data_wrapper.load_data()
data_ko = data_wrapper.load_kodata()#初始化并配置一个VGAE模型的训练器,用于在WT数据上训练模型
hyperparams = {"epochs": 100,
"lr": 7e-4,
"beta": 1e-4,
"seed": 8096}
log_dir=None
sensei = VGAE_trainer(data_wt,
epochs=hyperparams["epochs"],
lr=hyperparams["lr"],
log_dir=log_dir,
beta=hyperparams["beta"],
seed=hyperparams["seed"],
verbose=True,
)#模型训练
sensei.train()#保存训练模型
#sensei.save_model('model_example')# get distance between wt and ko
z_mu_wt, z_std_wt = sensei.get_latent_vars(data_wt)
z_mu_ko, z_std_ko = sensei.get_latent_vars(data_ko)
dis = gk.utils.get_distance(z_mu_ko, z_std_ko, z_mu_wt, z_std_wt, by="KL")
print(dis.shape)#根据扰动得分对基因进行排序,排名较前的说明敲除目标基因后收到的扰动越大
res_raw = utils.get_generank(data_wt, dis, rank=True)
res_raw.head()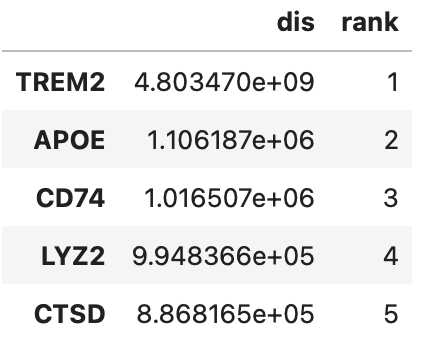
#permutation test
null = sensei.pmt(data_ko, n=100, by="KL")
res = utils.get_generank(data_wt, dis, null,
save_significant_as = 'MC_KOgenes'#将扰动基因保存到文件
)
res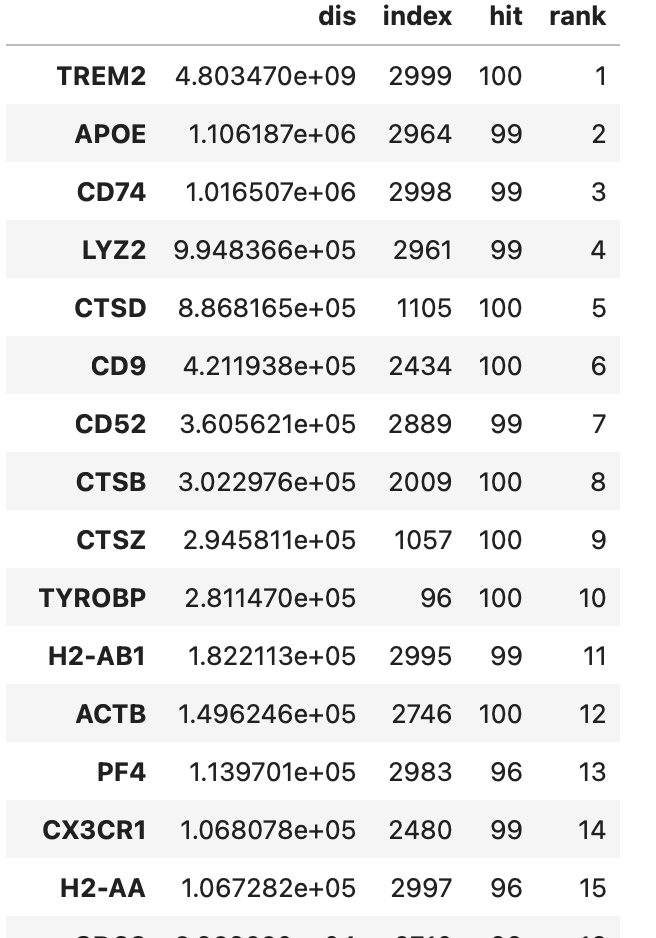
3、扰动基因富集分析及可视化
得到扰动基因列表,可以使用各种方式探究其功能,这里我们使用gseapy演示富集分析,参考我们之前的教程介绍:GSEApy【1】:广泛适用-python版差异基因富集分析
import pandas as pd
import gseapy as gp
import matplotlib.pyplot as pltgene_list = res.index.to_list()
enr = gp.enrichr(gene_list=gene_list, # gene形式需要与数据库保持一致
gene_sets=['GO_Biological_Process_2025','KEGG_2021_Human'],#需要分析的基因集,可以同时分析GO、KEGG两个gene set
organism='human', #物种
outdir=None)from gseapy import barplot, dotplot# categorical scatterplot
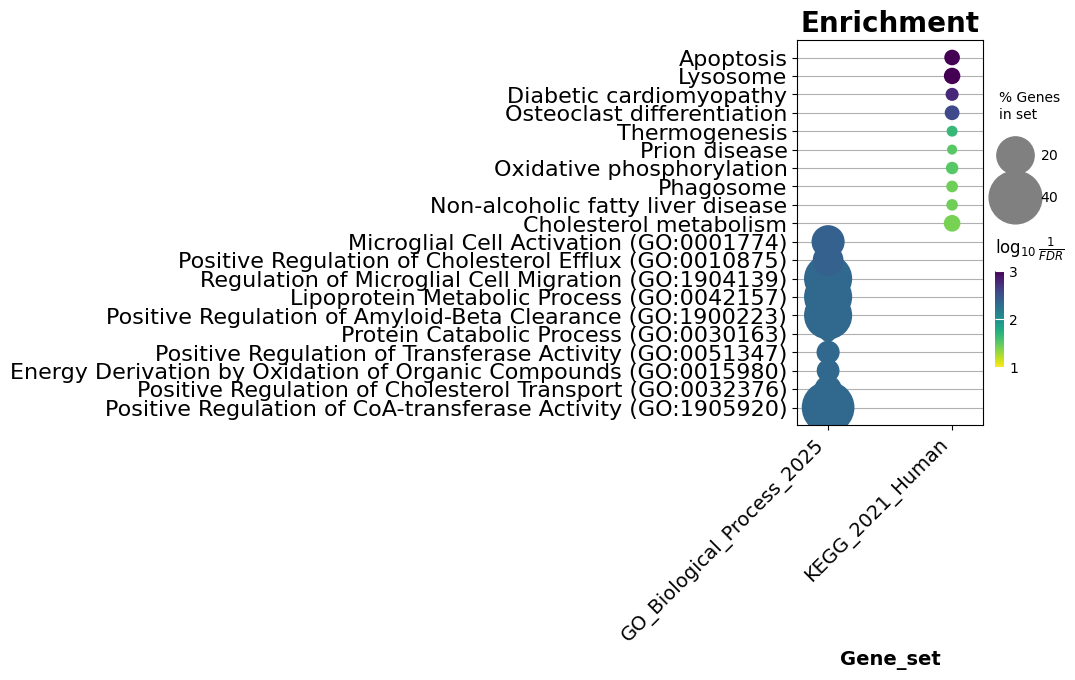
ax = dotplot(enr.results,#分析结果
column="Adjusted P-value",#按照adj pvalue展示结果
x='Gene_set', # set x axis, so you could do a multi-sample/library comparsion
size=10,#点大小
top_term=10,#展示的top5terms
figsize=(3,5),#图长宽
title = "Enrichment",#标题
xticklabels_rot=45, # rotate xtick labels
show_ring=False, # set to False to revmove outer ring
marker='o',#plot图形形式,参照https://matplotlib.org/stable/api/markers_api.html
)
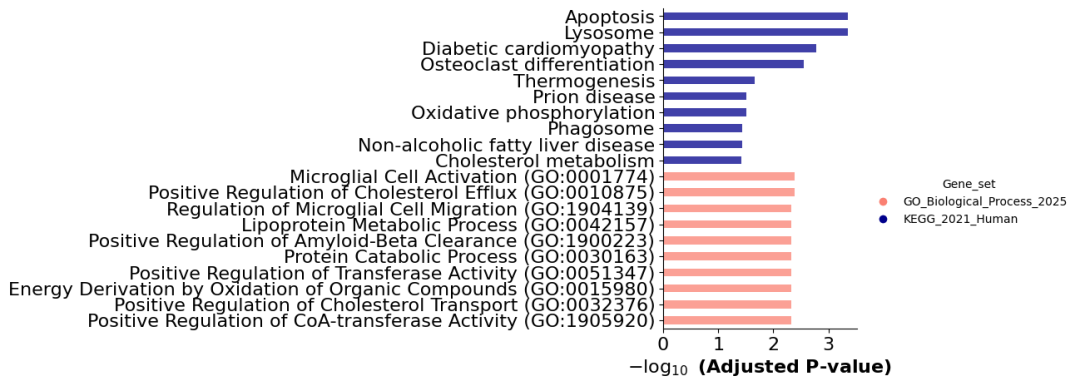
# categorical barplot
ax = barplot(enr.results,
column="Adjusted P-value",
group='Gene_set', # set group, so you could do a multi-sample/library comparsion
size=10,
top_term=10,
figsize=(3,5),
color = {'GO_Biological_Process_2025': 'salmon','KEGG_2021_Human':'darkblue'}
)
虚拟敲除本身就有一定的“虚拟”性,所以结果并不总是特别可靠,结合实际理性看待!觉得我们分享有用的点个赞再走呗!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号