PNAS | AI 蛋白结构预测的中间里程碑:trRosetta 方法全解析

PNAS | AI 蛋白结构预测的中间里程碑:trRosetta 方法全解析

DrugIntel
发布于 2026-07-08 16:53:57
发布于 2026-07-08 16:53:57

文献来源
论文题目:Improved protein structure prediction using predicted interresidue orientations 作者: Jianyi Yang, Ivan Anishchenko, Hahnbeom Park, Zhenling Peng, Sergey Ovchinnikov, David Baker 期刊: PNAS 发表时间: 2020 年 1 月 方法名称: trRosetta,transform-restrained Rosetta 代码链接: https://github.com/gjoni/trRosetta
导读
蛋白质结构预测长期依赖序列同源性、模板结构和共进化信号。CASP13 之后,深度学习已经能够从多序列比对中预测残基间距离分布,并将距离约束转化为三维结构。然而,距离只能告诉我们两个残基相隔多远,却不能完整描述它们在三维空间中的相对朝向。trRosetta 的核心贡献在于:在预测残基间距离之外,同时预测残基间取向信息,并把这些几何分布转化为 Rosetta 中的距离、二面角和平面角约束,通过快速能量最小化生成蛋白结构模型。该工作在 CASP13 和 CAMEO 回顾性 benchmark 上显著提升结构预测精度,并进一步显示模型可用于评估 de novo 设计蛋白的结构理想性。它不是 AlphaFold2 时代的终点,但它清楚展示了一个关键思想:蛋白结构预测真正需要学习的不是简单接触图,而是可直接约束三维折叠的残基间几何关系。
为什么这篇论文值得关注?
理解 trRosetta,最好不要把它只看成一个历史模型。它更像是 AlphaFold2 出现前,蛋白结构预测从接触预测走向几何约束预测的重要中间节点。
在早期共进化结构预测中,研究者通常关注两个残基是否接触。所谓接触,本质上是一个二分类问题:两个残基在三维结构中是否足够接近。这个信息很有用,因为在蛋白质序列进化过程中,如果两个残基在空间中相互接近,它们往往会发生协同突变,以维持结构稳定性或功能。
但接触图有一个明显问题:它过于粗糙。两个残基接触,并不等于它们在三维空间中的相对位置已经被确定。后来,CASP13 中 AlphaFold、RaptorX 等方法开始预测残基间距离分布,而不是简单预测接触与否。距离分布比接触图更连续、更丰富,可以更好地指导三维结构优化。
trRosetta 进一步推进了一步:距离还不够,还要预测取向。
两个残基之间的距离告诉你它们相隔多远,但不能告诉你它们如何相互朝向。对于蛋白折叠而言,取向信息非常关键,因为蛋白主链和侧链具有明确几何方向。只用距离约束折叠结构,可能仍存在大量满足距离但局部几何不合理的解;引入取向约束后,模型不只是把残基拉到合适距离,而是更明确地约束残基之间的相对空间变换。
这就是 trRosetta 的核心价值:它把深度学习从预测残基是否接触或相距多远,推进到预测残基之间更完整的局部刚体几何关系,并将这些预测转化为 Rosetta 可优化的结构约束。
论文中,trRosetta 由两个核心模块组成:
- 1. 一个从 MSA 输入预测残基间距离与取向的深度残差卷积网络;
- 2. 一个基于 Rosetta 的约束能量最小化建模流程。
前者负责从序列进化信息中学习几何约束,后者负责把这些约束转化为三维结构模型。
研究背景
蛋白质结构预测的基本问题是:给定一条氨基酸序列,预测它折叠后的三维结构。这个问题之所以困难,是因为序列到结构的映射高度非线性,且蛋白构象空间极其庞大。
在深度学习大规模进入该领域之前,主流方法大致有三类。
第一类是模板建模。如果目标蛋白在 PDB 中有同源结构,方法可以通过序列比对找到模板,再进行同源建模。它的优势是可靠、解释性强,但前提是有合适模板。对于没有可靠模板的新折叠蛋白,模板方法会明显受限。
第二类是片段组装与能量搜索,以 Rosetta 为代表。它依赖片段库、统计势函数和物理启发能量函数,在构象空间中搜索低能结构。问题在于,搜索空间巨大,能量函数不完美,尤其在缺乏远程约束时,很容易陷入错误折叠。
第三类是共进化约束建模。随着序列数据库增长,MSA 中可以提取残基协同变化信号,从而预测哪些残基在空间上接近。深度学习进一步把共进化特征映射为接触图或距离分布,使结构预测准确性大幅提高。CASP13 中,AlphaFold、RaptorX 等方法已经证明,预测残基间距离分布可以比传统接触预测提供更强的结构约束。
但 trRosetta 认为,距离预测仍然没有完全利用蛋白局部几何。蛋白结构不是一团点云,而是由具有方向性的主链和侧链组成。残基间距离只描述标量长度,无法完整描述残基局部坐标系之间的相对旋转关系。这就是本文切入的核心缺口。
论文的核心思想
trRosetta 的中心假设可以概括为:
如果深度网络不仅预测残基间距离,还预测残基间取向,并将这些几何分布转化为可优化的 Rosetta 约束,那么三维结构生成会更准确、更稳定。
这里有两个关键点。
第一个关键点是几何表示升级。论文不再只预测 Cβ-Cβ 距离,而是用 6 个参数描述两个残基之间的相对几何关系:
- • 距离 d;
- • 二面角 omega;
- • 二面角 theta12 和 theta21;
- • 平面角 phi12 和 phi21。
其中,omega 描述沿两个 Cβ 原子连线方向的旋转;theta 和 phi 描述一个残基的 Cβ 原子在另一个残基局部坐标系中的方向。由于 theta 和 phi 与残基顺序有关,所以它们是非对称的;d 和 omega 则是对称的。这 6 个量合在一起,可以更完整地确定两个残基主链原子之间的相对空间关系。
第二个关键点是建模流程不是端到端直接输出坐标。trRosetta 先用深度网络预测几何分布,再把这些分布转化为 Rosetta 的平滑能量势函数,作为结构优化约束。换句话说,深度学习负责从 MSA 中学习统计几何规律,Rosetta 负责在这些约束下搜索物理上更合理的三维结构。
这是一种非常典型的混合策略:学习模型提供信息丰富的约束,物理与统计能量函数负责结构生成和后处理。
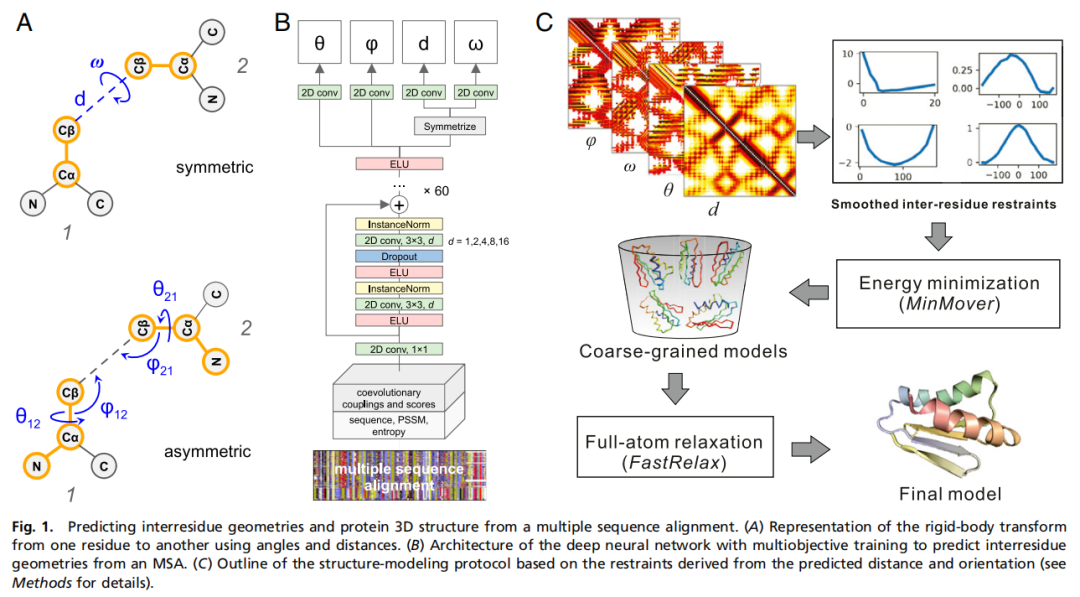
4. 方法细节:trRosetta 到底怎么做?
4.1 输入与输出
trRosetta 的输入是一条目标蛋白序列及其多序列比对 MSA。MSA 是整个方法的信息源,因为残基间共进化信号主要来自同源序列中的协同突变模式。
模型输出不是直接的三维坐标,而是所有残基对之间的几何概率分布,包括:
- • Cβ-Cβ 距离分布 d;
- • 二面角分布 omega;
- • 方向角分布 theta;
- • 平面角分布 phi。
这些概率分布随后被转化为 Rosetta 约束,用于生成三维结构。
所以完整流程是:
序列 → MSA → 共进化特征 → 残基间距离与取向分布 → Rosetta 约束势 → 粗粒度结构搜索 → 全原子松弛 → 最终结构模型
4.2 MSA 构建与选择
MSA 的质量直接影响预测结果。论文并没有只使用一个固定 MSA,而是为每个目标构建多个候选 MSA,再选择预测置信度更高的那个。
在测试阶段,作者为每个目标生成 5 种备选比对。前 4 种使用 HHblits 在 Uniclust30 数据库中搜索,分别设置不同 e-value cutoff:1e-40、1e-10、1e-3 和 1。最后一种采用逐步放宽 e-value 的迭代 HHblits 搜索,并在必要时使用 hmmsearch 搜索宏基因组序列数据库。这个宏基因组数据库包含约 70 亿条蛋白序列,来源包括 JGI metagenomes、metatranscriptomes、UniRef100、NCBI TSA 以及其他基因组资源。
为了避免过早引入过远同源序列或让 MSA 过深,作者设定了停止条件:当收集到至少 2000 条覆盖率 75% 的序列,或 5000 条覆盖率 50% 的序列时停止搜索。
这一步非常重要。论文中有一个失败例子 T1021s3-D2,完整序列 MSA 中大多数同源序列只覆盖第一个结构域,导致第二个结构域预测较差;使用结构域特异 MSA 后,TM-score 从 0.38 提升到 0.63。这个例子说明,trRosetta 的性能不只取决于网络,还高度依赖 MSA 是否正确覆盖目标结构区域。
4.3 输入特征:从 MSA 到 526 个通道
trRosetta 的网络输入是一个 L × L × 526 的二维张量,其中 L 是蛋白序列长度。
它的特征大致分为两类。
第一类是由单个位点特征转换来的二维特征。具体包括:
- • 查询序列的 one-hot 编码,20 个通道;
- • 位点氨基酸频率矩阵,20 种氨基酸加 gap,共 21 个通道;
- • 位点熵,1 个通道。
这些一维特征共 42 个通道。为了用于残基对预测,作者将其按行和列方向平铺,得到 84 个二维特征通道。
第二类是残基对共进化特征。作者从 MSA 中计算单点频率和双点频率,再构建协方差矩阵,并对经过收缩正则化的协方差矩阵求逆,得到残基对之间的耦合矩阵。每个残基对的耦合矩阵维度是 21 × 21,展平成 441 个通道。此外,作者还计算 Frobenius norm 并进行 APC 校正,得到 1 个额外通道。
因此,最终输入通道数为:
84 个平铺的一维特征 + 441 个耦合特征 + 1 个 APC 校正分数 = 526 个特征通道。
这个设计的意义在于,网络既能看到每个位置本身的氨基酸保守性,也能看到残基对之间的协同突变统计。
4.4 网络结构:扩张残差卷积 + 多任务几何预测
trRosetta 采用二维残差卷积网络处理 L × L 的残基对特征图。
网络首先用一个 1 × 1 卷积把输入通道从 526 降到 64。随后进入 61 个带扩张卷积的残差模块。扩张率按照 1、2、4、8、16 循环,这样网络既能捕捉局部残基对模式,也能扩大感受野,整合更长程的残基间依赖。论文图 1B 展示了这一架构:MSA 特征先进入二维卷积网络,经过多层残差模块后,在顶部分支预测不同几何目标。
输出层分为 4 个任务分支:
- • 距离 d 分布;
- • omega 角分布;
- • theta 角分布;
- • phi 角分布。
距离范围为 2–20 Å,被划分为 36 个 0.5 Å 的区间,另设一个非接触 bin。角度也被离散化为多个 bin:omega 和 theta 使用 15° 间隔,phi 使用对应的角度区间,并额外包含非接触 bin。每个分支最后通过 softmax 输出概率分布。
这里的设计很关键。trRosetta 不是训练多个完全独立的网络分别预测不同几何量,而是在主干网络中共享参数,只在最后分支输出不同目标。这样做的理由是,距离和取向并不是互相独立的任务。真实蛋白结构中,残基间距离、方向角、局部二级结构和长程拓扑存在强相关。共享网络可以让模型在学习距离时同时吸收取向信息,在学习取向时也利用距离约束。
4.5 训练目标:四个几何分布的分类交叉熵
trRosetta 把距离和角度预测都处理成分类问题。每个真实几何量落入一个离散 bin,网络输出该几何量属于各 bin 的概率分布。
训练损失为四个任务的 categorical cross-entropy 之和,四个任务权重相同。也就是说,模型被要求同时学会预测距离、omega、theta 和 phi。
训练集来自 PDB。作者先收集 2018 年 5 月 1 日前、X-ray 分辨率不低于 2.5 Å 的结构,提取长度至少 40 个残基的蛋白链,并按 30% 序列一致性去冗余。最终用于训练的是 15,051 条具有足够 MSA 同源序列的蛋白链。训练时还使用 MSA subsampling,即每轮训练随机抽取原始 MSA 的子集,使模型在不同深度和组成的 MSA 上学习,从而增强鲁棒性。
具体训练设置包括:长度超过 300 的蛋白随机裁剪到 300 残基以内,训练 100 个 epoch,Adam 优化器学习率 1e-4,L2 正则权重 1e-4,dropout keep probability 为 85%。作者训练了 5 个网络,采用随机 95/5 训练验证划分,最终预测取 5 个网络平均。单个网络在 NVIDIA Titan RTX GPU 上约需 9 天训练。
4.6 从概率分布到 Rosetta 约束势
trRosetta 最有方法学价值的部分,是如何把神经网络输出的离散几何分布变成 Rosetta 可以使用的结构约束。
对于每一对残基,网络输出的是距离和角度的概率分布。作者将这些概率分布转换为能量势函数。直觉上,如果某个距离或角度 bin 的预测概率高,那么结构优化时就应该偏好这个几何状态;如果概率低,则对应能量较高。
距离势函数采用类似 DFIRE 的思想,用最后一个距离 bin 作为参考态进行归一化;角度势函数则使用预测概率与非接触参考概率之间的对数比。所有离散分数随后通过 Rosetta 中的 spline 函数平滑,形成连续能量势。
不同几何量对应不同 Rosetta 约束类型:
- • 距离 d 转化为 AtomPair restraint;
- • omega 和 theta 转化为 Dihedral restraint;
- • phi 转化为 Angle restraint。
这样,神经网络预测的几何信息就从一张概率图变成了实际可以驱动折叠的能量项。
4.7 结构生成:粗粒度最小化 + 全原子松弛
结构生成分两阶段。
第一阶段是粗粒度 centroid 建模。蛋白主链保持全原子表示,但每个侧链用一个 centroid 原子近似表示。这样可以降低搜索复杂度。作者从随机主链二面角构象出发,用 Rosetta 的 MinMover 进行 quasi-Newton 能量最小化,优化算法为 L-BFGS。除了网络预测得到的距离和取向约束外,优化还加入 Rosetta centroid 能量项,包括 Ramachandran 约束、omega 角约束、van der Waals 排斥和主链氢键项。
为了降低陷入局部极小值的风险,作者引入两类随机性。
第一类是随机初始结构。每个目标尝试 10 个随机主链二面角起点。
第二类是不同约束集合。作者根据预测概率阈值筛选约束,阈值从 0.05 到 0.5,步长为 0.1;同时根据序列间隔设计三种约束加入策略:先短程再中程再长程、先短程加中程再长程、所有约束同时加入。
因此,每个目标共生成 150 个 centroid 模型,即 10 个初始结构 × 5 个概率阈值 × 3 种约束策略。随后选择能量较低的模型进入全原子松弛。全原子阶段使用 Rosetta FastRelax,并加入 ref2015 打分函数和约束项;最终选择总能量最低的全原子模型作为预测结构。
这个流程说明,trRosetta 并不是一个单纯神经网络模型,而是深度几何预测与 Rosetta 能量优化的结合。网络负责给出结构应满足的统计几何约束,Rosetta 负责在这些约束下生成物理上更可接受的结构。
5. 实验设计与关键结果
5.1 几何预测是否准确?
作者首先在 CASP13 的 31 个 free-modeling 目标上测试残基间几何预测。对于最高置信度的 7.5% 距离和取向预测,预测分布众数与真实结构之间具有较好相关性:距离 Pearson r 为 0.72,omega、theta、phi 的 circular correlation 分别为 0.62、0.77 和 0.60。更重要的是,top L 中长程接触的平均预测概率与真实 precision 相关性达到 r = 0.84。
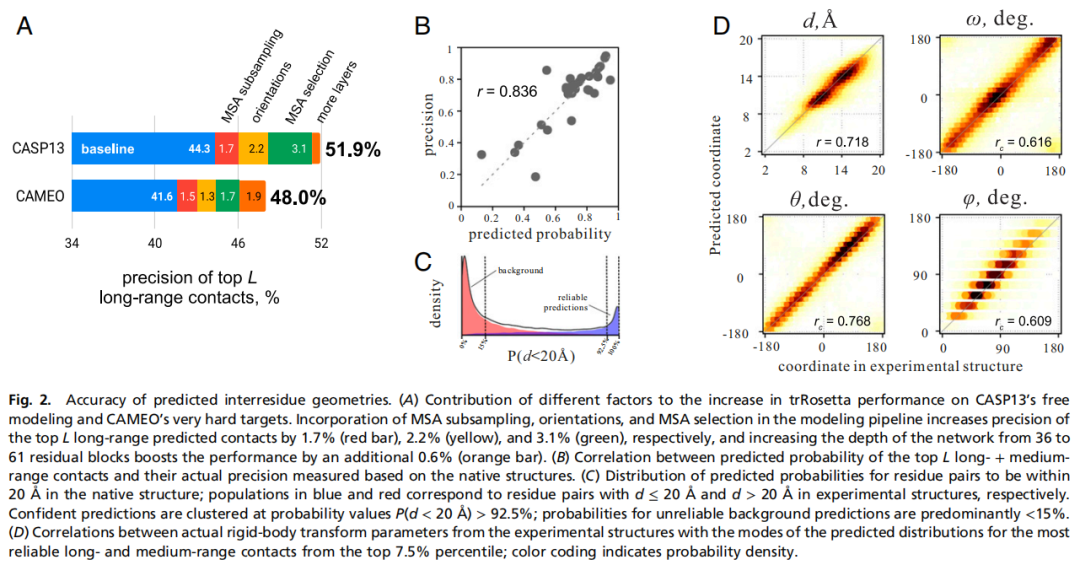
这说明网络输出的概率不仅能预测几何状态,也能反映预测可靠性。基于这个特性,作者可以在多个候选 MSA 中选择预测置信度最高的 MSA,进一步提升最终结构精度。
5.2 消融实验:哪些设计真正带来提升?
作者构建了一个只预测距离、没有 MSA subsampling 和 MSA selection 的 baseline 网络。与该 baseline 相比:
- • 加入 MSA subsampling,使 CASP13 FM top L 长程接触 precision 提升约 1.7%;
- • 加入取向预测,进一步提升约 2.2%;
- • 加入 MSA selection,再提升约 3.1%;
- • 加深网络后,总体比 baseline 提升约 7.6%。
在 CASP13 FM 上,trRosetta top L 长程接触 precision 达到 51.9%,高于 RaptorX-Contact 的 44.7% 和 TripleRes 的 42.3%;在 CAMEO very hard targets 上,trRosetta 长程接触 precision 为 48.0%,也高于 baseline 的 41.6%。
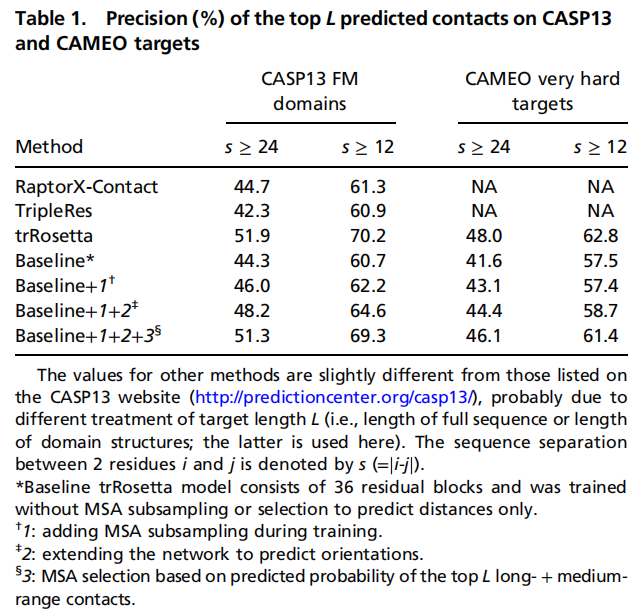
这个消融结果比较清楚地支持了作者的核心主张:提升不是单一技巧带来的,而是由 MSA 增强、取向预测、MSA 选择和网络深度共同贡献。其中,取向预测是方法概念上最重要的增量。
5.3 三维结构预测结果
在 CASP13 FM 目标上,trRosetta 平均 TM-score 为 0.625,高于 Zhang-Server 的 0.491,也高于 AlphaFold A7D 的 0.587。作者进一步拆解显示:baseline 距离预测驱动建模时 TM-score 为 0.537;使用完整网络的距离预测后提升到 0.592;加入取向分布后进一步提升到 0.625。
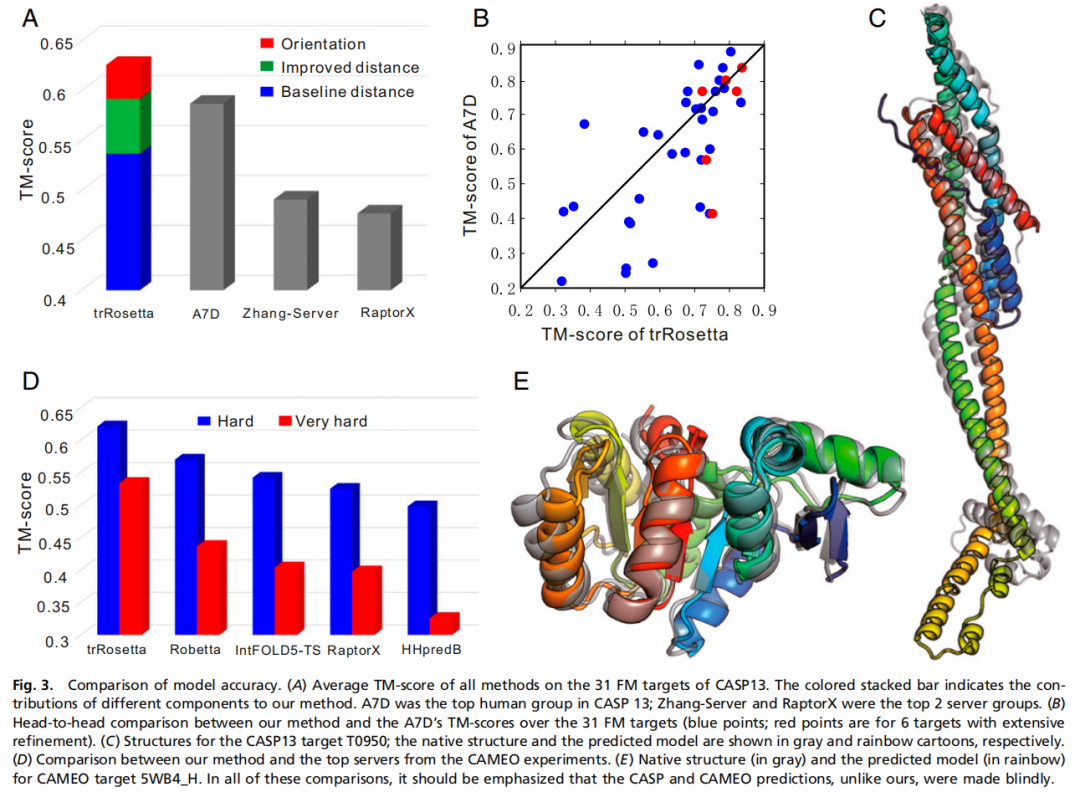
TM-score 衡量预测结构与真实结构的全局拓扑相似性,通常比 RMSD 更适合比较不同长度蛋白的整体折叠质量。trRosetta 的提升说明,取向约束不只是让接触图更准,也确实改善了最终三维结构。
作者还比较了结构生成协议本身的作用。使用 RaptorX-Contacts 的距离约束时,trRosetta 的 Rosetta 优化流程得到平均 TM-score 0.45,而 RaptorX 使用 CNS 建模得到 0.36。这提示结构生成端的优化策略也很重要,Rosetta 的采样和能量项有助于更好地满足预测约束。
在 CAMEO 131 个 hard targets 上,trRosetta 平均 TM-score 为 0.621,高于 Robetta 和 HHpredB。进一步筛选出更难的 66 个目标,即 HHpredB TM-score 小于 0.5 的目标后,trRosetta 平均 TM-score 为 0.534,比 Robetta 高 22%,比 HHpredB 高 63.8%。
5.4 距离和取向是否互补?
论文中一个很有启发性的结果是:距离和取向都可以单独驱动折叠,但二者组合效果最好。
在 CASP13 FM 目标上,仅用取向约束进行粗粒度建模,平均 TM-score 为 0.57;仅用距离约束为 0.55。经过松弛后,两者分别达到 0.58 和 0.59。虽然单独使用时差距不大,但结合距离和取向后模型质量进一步提高。
这说明取向信息不是距离信息的简单重复。距离决定残基对之间相隔多远,取向决定它们如何面对彼此。两者合并后,对蛋白折叠构象空间的约束更强。
5.5 de novo 蛋白设计:trRosetta 能否识别结构理想性?
论文最后一个亮点,是将 trRosetta 用于 de novo 设计蛋白的结构评估。
作者收集了 18 个具有实验结构的 de novo 设计蛋白,包括 alpha、beta 和 alpha/beta 拓扑,并选取 79 个长度相近的天然蛋白作为对照。值得注意的是,de novo 设计蛋白通常没有天然同源序列,因此几乎没有传统意义上的共进化信号。尽管如此,trRosetta 仍能从单条设计序列中准确预测这些设计蛋白的结构,预测模型与晶体结构高度接近。
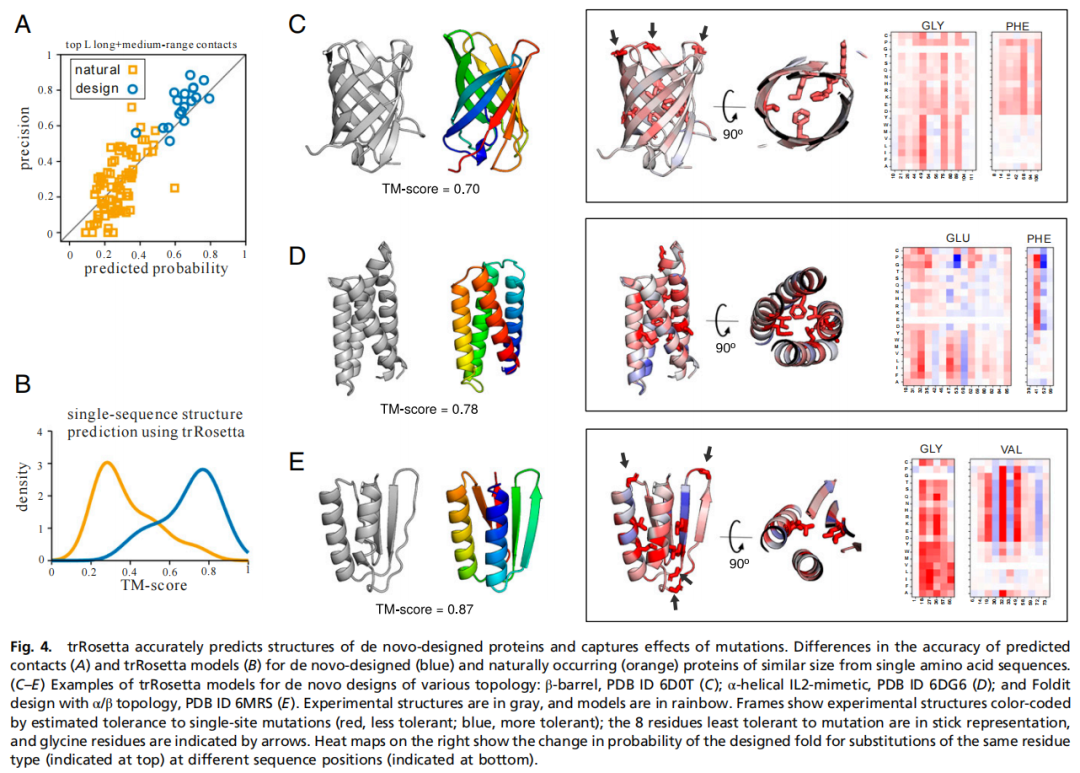
这说明模型并不只是机械读取 MSA 共进化信号,还学到了一部分序列到结构的普遍规律。作者进一步做了单点突变扫描:对设计蛋白每个位置逐一替换氨基酸,并计算突变后 top L 中长程接触概率的下降程度。结果显示,核心疏水残基以及 beta-turn 中的 glycine 突变会显著降低设计结构概率;同一种氨基酸替换在不同位置的影响差异很大,说明模型捕捉到明显的结构上下文依赖,而不是只依赖 BLOSUM 或 PAM 这类平均替换矩阵。
这部分结果对蛋白设计很重要。它提示 trRosetta 可以作为设计序列的结构合理性评估器,用来判断一个设计是否符合稳定折叠的几何规律,也可以帮助识别对折叠最关键的残基。
启发
启发一:蛋白结构预测的核心不是接触,而是几何
接触图只是最粗粒度的结构约束。距离分布已经更进一步,但仍缺少方向信息。trRosetta 的关键贡献是把预测目标从残基是否接近,推进到残基对之间的局部刚体几何关系。这一思想后来也成为蛋白结构预测模型发展的重要方向:模型需要学习的是三维空间中的约束,而不是仅仅学习二维接触。
启发二:深度学习和物理建模并不是替代关系
trRosetta 没有让神经网络直接输出最终坐标,而是把深度学习输出转化为 Rosetta 约束,再通过能量最小化生成结构。这种混合建模在 AI for Science 中很有代表性:学习模型从数据中提取复杂统计规律,物理或几何优化负责保证结构连续性和合理性。
启发三:预测置信度本身也是有价值的信息
trRosetta 发现预测概率与真实接触 precision 高度相关,因此可以利用预测概率进行 MSA 选择和模型质量估计。这一点对 AI 制药也很重要:模型不仅要给答案,还要给出可用的置信度信号,否则很难进入真实决策流程。
启发四:蛋白设计需要可解释的结构评估器
在 de novo 设计蛋白中,trRosetta 对设计结构给出更高概率,并能通过突变扫描识别关键折叠残基。这提示结构预测模型不只是预测工具,也可以成为蛋白设计中的判别器、过滤器和突变效应评估器。
局限性
1. 方法仍然强依赖 MSA 质量
trRosetta 的主要信息来源仍是 MSA。对于天然蛋白,如果同源序列不足、MSA 覆盖不均、结构域边界不清,预测可能明显下降。T1021s3-D2 的例子已经说明,错误或不完整的 MSA 会直接影响结构质量。
2. 对蛋白动态、复合物和功能环境考虑有限
本文主要关注单链蛋白结构预测。真实生物体系中,蛋白可能存在构象变化、无序区域、膜环境、配体诱导适配、复合物装配、翻译后修饰等因素。trRosetta 的静态结构预测不能直接解决这些问题。
3. 结构精度不等于功能预测能力
TM-score 提高说明整体折叠更接近真实结构,但并不保证活性位点几何、结合界面、催化残基排布或动态构象状态都正确。对于 AI 制药而言,整体结构预测只是第一步,后续还需要口袋构象、配体结合、蛋白柔性和热力学稳定性等更细粒度验证。
4. de novo 设计结果有启发性,但不是充分验证
trRosetta 能更准确预测 18 个 de novo 设计蛋白,并从单序列中识别折叠关键残基,这很有意义。但该实验并不等于模型已经可以广泛指导所有蛋白设计。设计蛋白往往更规则、更理想化,可能比天然蛋白更接近模型学到的稳定结构规律。对于复杂功能蛋白、酶活性设计或结合界面设计,还需要更多实验验证。
对 AI 制药未来发展的意义
trRosetta 对 AI 制药的价值,不在于它直接解决药物发现问题,而在于它提供了一种非常清晰的建模思想。
第一,结构预测可以服务于靶点建模。对于缺乏实验结构的靶点,可靠结构模型可以支持口袋识别、突变解释、功能区域分析和结构基础虚拟筛选。虽然今天 AlphaFold2、AlphaFold3 等模型已经大幅推进这一方向,但 trRosetta 展示的几何约束思想仍然具有方法学价值。
第二,残基间几何分布可以成为结构设计的评价信号。蛋白设计不是只生成序列,而是要生成能够稳定折叠并具有功能的序列。trRosetta 对 de novo 设计蛋白的分析表明,结构预测模型可以用于判断设计序列是否符合稳定折叠规则,并定位关键残基。
第三,AI 制药中的很多问题都需要从标量预测走向几何预测。无论是蛋白结构预测、分子对接、蛋白-配体复合物建模,还是蛋白-蛋白相互作用预测,核心都不是简单预测一个分数,而是预测可解释、可优化、可约束的三维几何关系。trRosetta 用残基间距离和取向连接深度学习与 Rosetta 优化,这种思想对后来的结构生成、扩散模型和几何深度学习都有启发。
第四,模型置信度需要进入实际决策流程。trRosetta 将预测概率用于 MSA 选择和模型准确性估计,说明一个模型如果能提供可靠置信度,就更容易被用于真实科研流程。AI 制药中的候选分子生成、对接打分和蛋白设计同样需要这种可靠性估计。
总结与推荐理由
trRosetta 的贡献可以用一句话概括:
它把蛋白结构预测中的深度学习输出,从残基间距离推进到残基间完整几何取向,并通过 Rosetta 约束优化把这些预测转化为三维结构模型。
这篇论文值得读,不是因为它在今天仍然代表最高结构预测性能,而是因为它清楚展示了结构生物学 AI 发展的一个关键方向:模型需要学习能直接约束三维结构的几何关系,而不只是学习接触图或整体评分。
对于关注 AI 蛋白结构预测、蛋白设计和 AI 制药基础模型的读者,trRosetta 是理解从共进化接触预测走向几何深度学习的重要文献,也是理解 AlphaFold2 前夜技术路线演化的一块关键拼图。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号