被AI打败的系列:复现Nature图表转为学习可视化思路

被AI打败的系列:复现Nature图表转为学习可视化思路

KS科研分享与服务-TS的美梦
发布于 2026-07-08 20:09:27
发布于 2026-07-08 20:09:27
之前我们有一个系列,小伙伴遇到文章中比较好的可视化的时候总会让我们进行复现,**但这个系列已经被AI打败了**,我们也很久没有再出过类似的教程了。结合以前的风格,在复现的前提下,力求创新和“为我所用”,所以我想复现系列改为学习可视化思路更好。使用AI实现本来的面目,然后利用自己的数据进行修饰。**可视化的重点在于思维,还在你的脑海中,AI目前也只能实现你的想法,并不能自主思考**。
今天带来的图表是发表于Nature正刊,Schwarz, L.A., Dotter, C.P., Isaev, S. et al. Cortical development dynamics across autism spectrum disorder mouse models. Nature (2026). https://doi.org/10.1038/s41586-026-10679-1。原文展示的是celltype丰度数据,关于celltype abundance参考(你有这么大的样本吗?---学习张泽民院士Cell文章分析思路之单细胞丰度NMF分析,miloR:单细胞差异丰度分析,milo-基于KNN的差异丰度分析(python版miloR),单细胞组织偏好性Ro/e指数计算方法统一测试及可视化)等等。图中直接用三角形展示了上下变化关系,并用不同颜色展示p值,三角形大小代表丰度。
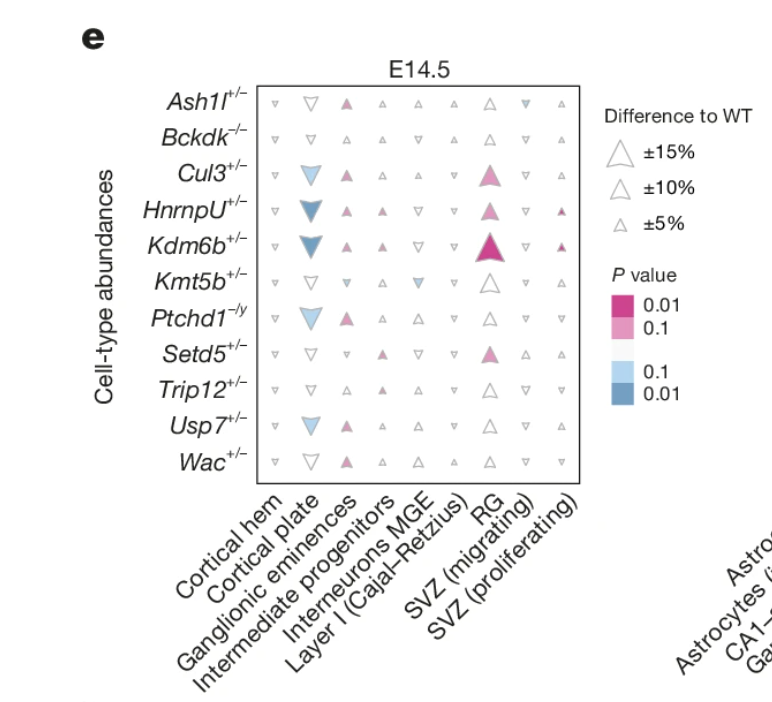
我直接把这幅图给了GPT,让它使用R语言实现,没有更多的信息,很快它按照图创造了示例数据,并给出了代码,代码直接就可以运行。当然,如果你使用codex,给它你的数据,可能会进行整理,按照这个风格可视化。如果有更详细的信息需求输入,会得到更好的结果。
library(tidyverse)
library(dplyr)
# 构造一个示例数据框:行=基因型,列=细胞类型
set.seed(1)
gene <- c("Ashd1+/-", "Bsdk+/-", "Cul3+/-", "Hnrnpu+/-",
"Kdm6b+/-", "Pctd5+/-", "Setd1b+/-", "Usp7+/-", "Trip12+/-", "Wac+/-")
cell_type <- c("Cortical plate",
"Intermediate progenitors",
"Ganglionic eminence",
"Layer (Cajal-Retzius)",
"SVZ (migrating)",
"SVZ (proliferating)")
# 造一份长数据:一行一个 (gene, cell_type)
df <- expand.grid(gene = gene, cell_type = cell_type) %>%
as_tibble() %>%
mutate(
diff_to_WT = rnorm(n(), mean = 0, sd = 0.08), # -0.2 ~ 0.2 之间的随机差异 (比例,负表示减少)
diff_pct = diff_to_WT * 100, # 转成百分比
pvalue = runif(n(), min = 0, max = 0.1) # 0~0.1 之间的随机 p 值
)
df_plot <- df %>%
mutate(
# 上/下三角:diff_to_WT > 0 用 shape 24 (上三角),<0 用 shape 25 (下三角)
shape_tri = ifelse(diff_to_WT >= 0, 24, 25),
# 大小:用绝对值的百分比;你可以据情况缩放
size_abs = abs(diff_pct),
# p 值分箱:与你图例类似(0.01、0.1 等)
p_bin = case_when(
pvalue <= 0.01 ~ "p ≤ 0.01",
pvalue <= 0.05 ~ "0.01 < p ≤ 0.05",
pvalue <= 0.1 ~ "0.05 < p ≤ 0.1",
TRUE ~ "p > 0.1"
)
)
# df_plot <- df_plot %>%
# mutate(
# gene = factor(gene, levels = rev(gene)), # 从上到下(你也可以手动按论文顺序写 levels)
# cell_type = factor(cell_type, levels = cell_type) # 从左到右
# )
library(ggplot2)
ggplot(df_plot, aes(x = cell_type, y = gene)) +
geom_point(
aes(
size = size_abs,
fill = p_bin,
shape = factor(shape_tri)
),
color = "black", # 三角的边框颜色
alpha = 0.9
) +
scale_shape_manual(
values = c("24" = 24, "25" = 25),
guide = "none" # 不需要单独显示 shape 图例(只看颜色和大小)
) +
# 大小:根据你的数据范围调试 range
scale_size_continuous(
range = c(1, 8),
breaks = c(5, 10, 20), # 例如 5%、10%、20% 差异
name = "Difference to WT (%)"
) +
# 填充颜色:接近原图的蓝-粉色系(示例)
scale_fill_manual(
values = c(
"p ≤ 0.01" = "#b2182b",
"0.01 < p ≤ 0.05" = "#ef8a62",
"0.05 < p ≤ 0.1" = "#67a9cf",
"p > 0.1" = "#cccccc"
),
name = "P value"
) +
theme_minimal(base_size = 12) +
theme(
axis.title.x = element_blank(),
axis.title.y = element_blank(),
axis.text.x = element_text(angle = 45, hjust = 1),
panel.grid.major = element_blank(),
panel.grid.minor = element_blank()
) +
coord_cartesian(clip = "off")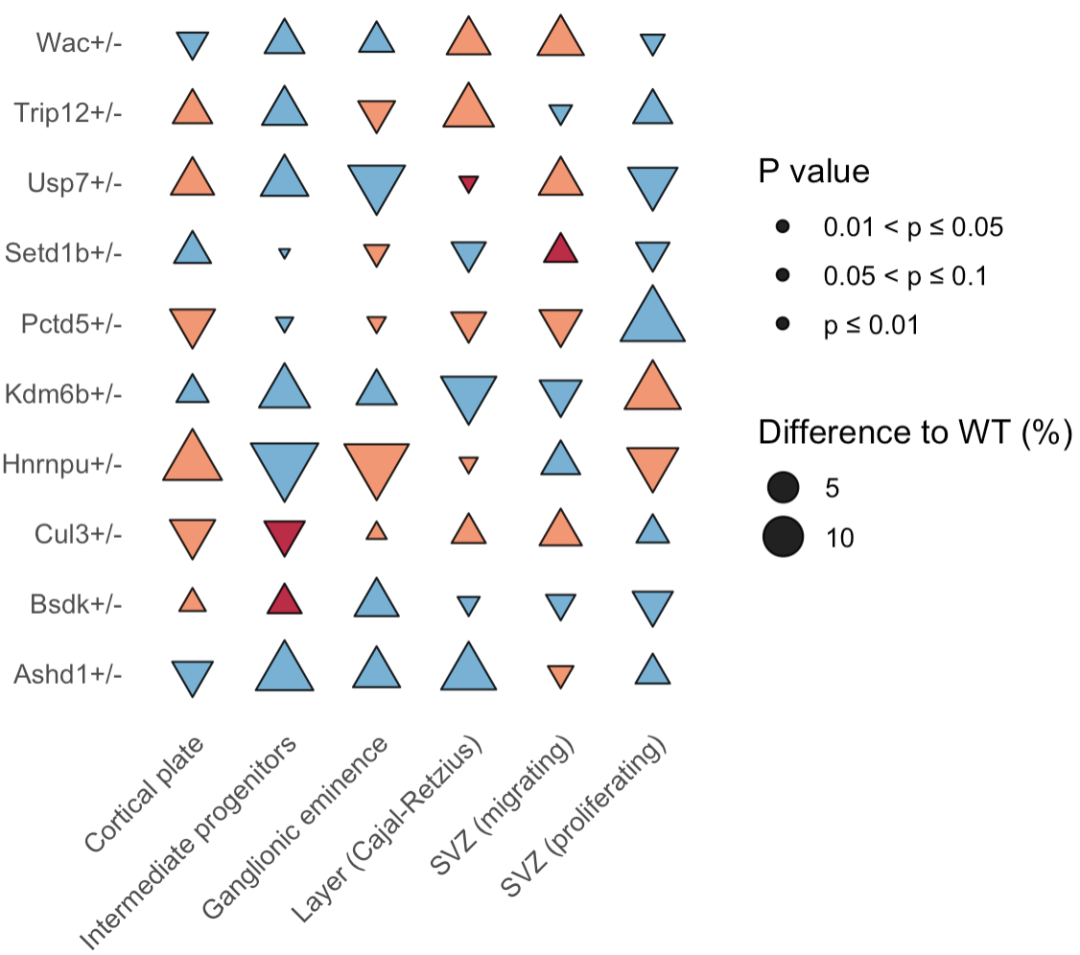
这个图还可以应用到哪里?可以是同时展示目的基因集在多种celltype中的上下调关系。至于还有哪些用处和修饰,还是需要动动自己聪明的小脑壳了。AI会给你提供方案,大大提高效率,但是要创造出新的内容,还需要自己。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-08,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号