NeurIPS | RGFN:在化学反应空间中生成可合成小分子

NeurIPS | RGFN:在化学反应空间中生成可合成小分子

DrugIntel
发布于 2026-03-30 16:08:12
发布于 2026-03-30 16:08:12

论文信息
- • 标题:RGFN: Synthesizable Molecular Generation Using GFlowNets
- • 会议:NeurIPS 2024
- • 机构:Mila / 蒙特利尔大学、多伦多大学、雅盖隆大学、麦吉尔大学、多伦多病童医院、Acceleration Consortium
- • 通讯作者:Michał Koziarski、Andrei Rekesh、Dmytro Shevchuk(共同一作)
- • 代码:https://github.com/koziarskilab/RGFN
目录
- 1. 研究背景与核心动机
- 2. 相关工作梳理
- 3. GFlowNet 理论基础
- 4. RGFN 方法详解
- • 4.1 整体框架与生成流程
- • 4.2 前向策略各阶段的概率建模
- • 4.3 后向策略设计
- • 4.4 动作嵌入:分子指纹扩展
- 5. 化学语言的设计与状态空间估计
- 6. 实验设计
- • 6.1 评测任务与 Oracle
- • 6.2 GPU 加速对接的引入
- • 6.3 基线方法
- • 6.4 超参数与训练细节
- 7. 实验结果分析
- • 7.1 奖励分布对比
- • 7.2 模式发现能力(Modes)
- • 7.3 合成可行性评估
- • 7.4 片段库规模扩展实验
- • 7.5 生成配体的结构多样性与对接姿态
- • 7.6 成本分析
- 8. 局限性深度讨论
- 9. 对药物发现领域的启示
1. 研究背景与核心动机
1.1 药物类分子空间的巨大规模
类药分子(drug-like molecules)的理论数量估计约为 ,这一数字远超宇宙中原子的数目,使得"对全空间进行穷举筛选"在物理上是不可能的。传统基于实验的高通量筛选(HTS)和基于预先构建的虚拟化合物库(如 Enamine REAL,约 65 亿化合物)的计算筛选,仅能覆盖这一巨大空间的极小角落。
1.2 生成模型的崛起与核心缺陷
生成式机器学习方法通过直接从目标性质分布中采样来规避穷举问题,近年来涌现出大量方法:
- • SMILES 序列生成(如 RNN、Transformer)
- • 分子图生成(如 JTVAE、GraphGA)
- • 三维坐标生成(如 diffusion-based 方法)
- • GFlowNet 框架(如 FGFN)
然而,这些方法普遍面临一个根本性矛盾:优化目标(高活性/高评分)与合成可行性之间缺乏内在约束。大量研究表明,主流生成模型产出的"高分"分子中,相当比例在化学合成上要么极为困难,要么完全不可行。
这一问题的严重性在于:
- 1. 实验验证无从开展:无法合成的分子无法进行生物活性测试。
- 2. 资源浪费:计算资源和化学家时间被用于"分析不可能的化合物"。
- 3. 过拟合风险:模型可能通过生成"化学不可能"的结构来"欺骗"评分函数。
1.3 现有应对策略的不足
策略 | 代表方法 | 局限性 |
|---|---|---|
事后过滤(SAScore, SYBA) | FGFN+SA | 启发式评分噪声大,无法保证合成可行性 |
逆合成路径预测 | AiZynthFinder, RetroGNN | 逆合成模型本身精度有限,泛化困难 |
直接在反应空间操作 | SyntheMol, casVAE, RGFN | 搜索空间设计和扩展性是关键 |
RGFN 属于第三类策略中最新的代表性工作,其核心思想是:将分子生成过程与分子合成过程统一起来——分子不是被"设计出来"的,而是被"合成出来"的。
2. 相关工作梳理
2.1 分子生成方法分类
基于表示的分类
表示形式 | 典型方法 | 优势 | 劣势 |
|---|---|---|---|
SMILES | RNN、Transformer | 简单高效 | 有效性约束弱 |
分子图 | JTVAE, GraphGA | 可保证分子有效性 | 不保证合成性 |
3D 坐标 | DiffSBDD, SILVR | 结构感知 | 计算成本高 |
反应树 | casVAE, RGFN | 天然保证合成性 | 搜索空间受限 |
基于方法论的分类
- • 变分自编码器(VAE):JTVAE, Mol-CycleGAN
- • 强化学习(RL):REINVENT, GCPN
- • 遗传算法(GA):GraphGA, GuacaMol
- • 扩散模型:DiffSBDD, SILVR
- • GFlowNet:FGFN, RGFN, TacoGFN, SynFlowNet
2.2 合成可行性相关工作
- • SAScore(Ertl & Schuffenhauer, 2009):基于片段贡献的启发式评分,计算快但精度有限。
- • AiZynthFinder(Genheden et al., 2020):基于机器学习的逆合成规划工具,可找到有效合成路径,但依赖预训练数据分布,对新颖结构泛化差。
- • SyntheMol(Swanson et al., 2024, NMI):基于 Monte Carlo 树搜索,在 Enamine REAL 反应和构建块空间中搜索抗菌候选物,但构建块成本较高,生成速度慢。
- • casVAE(Nguyen & Tsuda, 2022):通过级联 VAE 生成反应树,但采样效率低,计算开销大。
- • SynFlowNet(Cretu et al., 2024, ICLR Workshop):与 RGFN 同期的工作,同样将 GFlowNet 应用于合成路径空间,但未公开代码和精选反应/构建块库。
- • RetroGFN(Gainski et al., 2024):本团队的另一工作,将 GFlowNet 用于多样可行的逆合成规划。
3. GFlowNet 理论基础
3.1 核心定义
GFlowNet 定义在一个有向无环图(DAG) 上,其中:
- • 为状态集合,包含唯一起始状态 (无入边)和终止状态集合 (无出边);
- • 为动作(边)集合, 表示从状态 执行动作后转移至 ;
- • 奖励函数 对终止状态(即最终生成的分子)给出正值奖励。
目标:训练一个前向策略 ,使得从 出发采样的终止状态 满足:
即高奖励分子被更频繁采样,同时保持多样性(不坍缩到单一最优解)。
3.2 流匹配条件
对于非负流函数 ,完美训练的 GFlowNet 应满足全局流匹配约束:
以及终止状态条件:。
3.3 轨迹平衡(Trajectory Balance)训练目标
RGFN 采用轨迹平衡(TB)损失(Malkin et al., 2022, NeurIPS),这是目前 GFlowNet 中信用分配效果最好的训练目标之一。
对于完整轨迹 ,TB 要求以下等式成立:
其中:
- • :可学习的标量,近似配分函数;
- • :前向策略(生成分子所用);
- • :后向策略(用于信用分配,可视为"分子 是如何一步步被拆解为其构成组件的")。
训练损失为对所有采样轨迹的上式两侧取对的均方误差(MSE):
3.4 与强化学习的关键区别
强化学习(RL)通常优化期望奖励 ,在稀疏奖励情况下容易坍缩为单一模式(Mode Collapse)。GFlowNet 的采样分布与奖励成正比,天然具有探索多样模式的能力,在药物发现这类需要发现"尽可能多的不同高活性骨架"的任务中,理论上优于 RL。
4. RGFN 方法详解
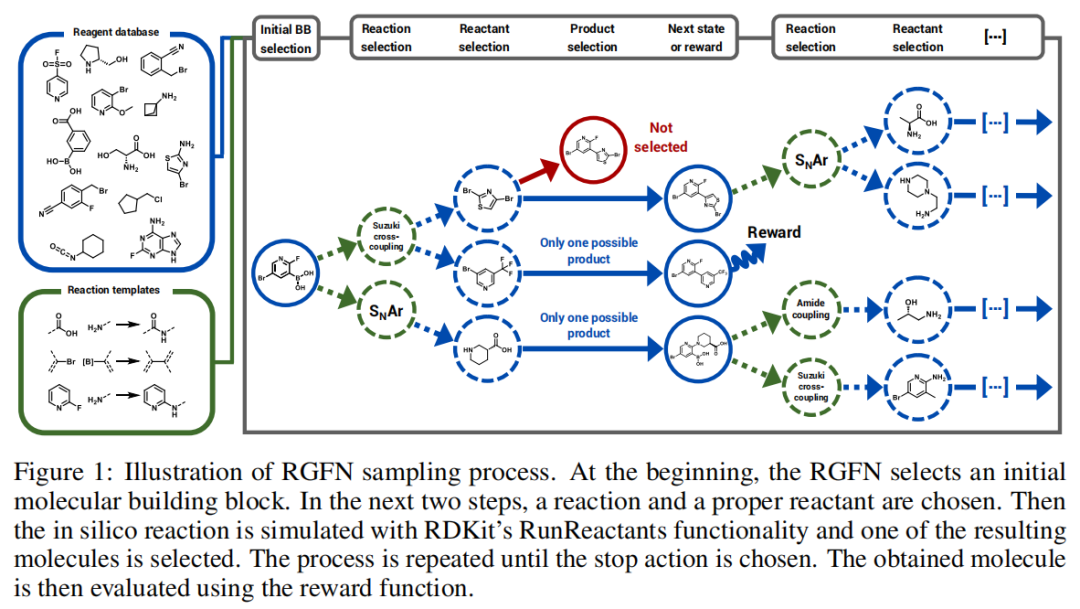
4.1 整体框架与生成流程
RGFN 将分子生成过程建模为一系列化学步骤的序列,每一步对应一次真实的化学反应。完整的生成轨迹包括:
s₀(空分子)
└─[选构建块]→ m₀(初始原料)
└─[选反应 r₁]→ [选第二原料 m₁]→ [执行反应,选产物]→ m₁'
└─[选反应 r₂]→ [选第三原料 m₂]→ [执行反应,选产物]→ m₂'
└─...─→ [停止动作] → 最终分子 x
↓
计算 R(x)底层化学反应使用 RDKit 的 RunReactants 函数在 Python 中模拟执行,反应规则编码为 SMARTS 模板。
前向策略的骨干网络为图Transformer(Graph Transformer, Yun et al., 2019, NeurIPS),接受分子图输入,输出固定维度的分子嵌入 (包括空图 ),并支持以反应 为条件的嵌入 。
4.2 前向策略各阶段的概率建模
阶段一:选择初始构建块
模型从空图出发,通过一个 MLP 对全体构建块打分,经 softmax 归一化得到选择概率。
阶段二:选择反应模板
输出维度为 ,额外一维对应"停止"动作。对于在当前分子上无法应用的反应,其 logit 强制设为 (硬过滤)。
阶段三:选择第二个反应物
注意 与阶段一共享参数,通过将当前分子状态 与反应 共同编码,实现对"在当前中间体上,用反应 所需的最佳搭档原料"的预测。不兼容的构建块被硬过滤。
阶段四:执行反应并选择产物
执行反应后可能产生多个化学上合法的产物(由于反应位点的多重匹配)。从产物集合 中选择产物:
这里每个候选产物单独经过图变换器嵌入,再打分排序。
4.3 后向策略设计
后向策略 刻画的是:给定当前分子 ,它是由哪个前驱状态(, 反应 , 反应物 )经过一步反应得到的?
设 为所有可能产生 的三元组 的集合(需递归验证 能在 步内合成),则:
后向策略使用独立的图变换器骨干网络 (结构与前向相同但参数不共享)。
设计要点:后向策略对于 TB 损失的有效性至关重要——它需要正确表征"分子 可以通过多少条不同合成路径得到",从而在信用分配时对多路径分子给予恰当的奖励分配。
4.4 动作嵌入:分子指纹扩展
问题动机
当构建块库从 350 扩展到数千甚至数万时, 的标准实现(将每个构建块视为独立 index,通过 softmax 打分)面临严重的扩展性问题:
- 1. 模型无法感知构建块之间的结构相似性(即选择 的梯度不会影响结构相似的 );
- 2. 随着库的增大,有效搜索所需的训练步骤数急剧增加。
解决方案:MACCS 指纹嵌入
将构建块选择的 logit 计算从独立嵌入改为基于分子指纹的内积相似度:
其中:
- • :当前中间体在反应 条件下的图变换器嵌入(维度 );
- • :可学习线性投影矩阵;
- • :GELU 激活函数;
- • :构建块 的嵌入,由 MACCS 166 位分子指纹 线性映射加上 index 嵌入拼接得到。
关键优势
- • 结构相似的构建块获得相近的嵌入 ,使得梯度能在结构相似的构建块之间有效传播;
- • 所有构建块的 可以预先计算并缓存,推理时无额外计算开销;
- • 当 退化为纯 index 嵌入时,该公式等价于标准 MLP 实现,保持了向下兼容性。
5. 化学语言的设计与状态空间估计
5.1 反应类型(17 大类,132 个 SMARTS 模板)
RGFN 选择的反应类型均满足:工业和学术界广泛使用、条件温和、收率高(75–100%)、原料来源广泛:
类别 | 反应名称 | 类型 |
|---|---|---|
C–N 键形成 | 酰胺键形成(Amide coupling) | 普通反应 |
C–N 键形成 | 亲核芳香取代(SNAr) | 普通反应 |
C–C 键形成 | Suzuki-宫浦偶联 | 过渡金属催化 |
C–N/C–O 键 | Buchwald-Hartwig 偶联 | 过渡金属催化 |
C–C 键 | Sonogashira 偶联 | 过渡金属催化 |
C–C 键 | Michael 加成 | 亲核加成 |
C–N–C=O | 脲合成(Isocyanate route) | 普通反应 |
C–N–C=O | 脲合成(Carbonyl surrogate) | 隐式反应 |
S–F 键 | 磺酰氟交换(SuFEx) | Click 化学 |
S–N 键 | 磺酰氯取代 | 普通反应 |
杂环合成 | 叠氮-炔烃环加成(CuAAC) | Click 化学 |
杂环合成 | 叠氮-腈环加成 | 隐式反应 |
酯类 | 酯化反应 | 普通反应 |
酰胺还原 | 酰胺还原为胺 | 官能团变换 |
杂环合成 | 肽末端硫脲环化(→ iminohydantoin) | 普通反应 |
杂环合成 | 肽末端硫脲环化(→ tetrazole) | 隐式反应 |
隐式反应(Implicit Reactions):反应产物含有 SMARTS 中未明确指定的原子,这些原子来自"隐式试剂"(如脲合成中的碳酰二咪唑 CDI,不需要在模板中显式编码)。
代理反应(Surrogate Reactions):由于部分对接软件(QuickVina2)不支持含硼原子的配体,硼酸基团用含 C 标记的羧酸进行等排替换,同时保留原有 SMARTS 兼容性。
考虑到反应物顺序交换的双向版本,132 个 SMARTS 模板覆盖了全部 17 类反应的所有变体。
5.2 构建块库(350 个低成本原料)
筛选标准:
- • 价格 ≤ (实际平均约22.52/g,最低 ,最高190/g);
- • 与预定义 17 类反应中至少一类兼容;
- • 商业可及(来自 Sigma-Aldrich, Oakwood Chemicals, Combi-Blocks, TCI, AngeneSci 等主流供应商)。
分组平衡:为防止引入学习偏差,构建块按其在反应中的角色被分为 12 个组(如羧酸类、胺类、芳基硼酸类等),并根据各组在不同反应中的出现频率赋予权重系数,使用公式:
进行比例采样(Michael 受体组权重特别低 = 0.25,因其结构多样性受限)。
5.3 状态空间规模估计
直接计算 RGFN 可生成的分子总数是不可行的(DAG 结构过于复杂),因此采用蒙特卡洛估计法:
基于 1000 条随机轨迹的统计结果(见原文 Table 2):
最大反应步数 | RGFN (350) 估计规模 | RGFN (8350) 估计规模 | Enamine REAL |
|---|---|---|---|
0(纯构建块) | ~350 | ~8317 | — |
2 | ~ | ~ | — |
3 | ~ | ~ | — |
4 | ~ | ~ | 6.5 × |
5 | ~ | ~ | — |
关键结论:即使仅用 350 个低成本构建块,最多执行 4 步反应,RGFN 的搜索空间已比 Enamine REAL 大约 倍;使用 8350 个构建块时,差距扩大至 倍以上。
6. 实验设计
6.1 评测任务与 Oracle
论文使用了六个不同的评测任务,覆盖了从简单代理模型到真实物理计算的完整谱系:
任务 | 类型 | Oracle 模型 | 备注 |
|---|---|---|---|
sEH 亲和力 | 代理模型 | MPNN(训练于归一化对接分数) | 常用基准,来自 Bengio et al., 2021 |
Senolytic 活性 | 代理模型 | GNEprop(5层 GIN + JK) | 极不平衡数据集(<100 个活性分子),稀疏奖励 |
DRD2 活性 | 代理模型 | SVM + ECFP6 核 | 多巴胺 D2 受体,来自 TDC |
ClpP 对接 | 直接对接 | Vina-GPU 2.1(GPU 加速) | ATP 依赖性蛋白酶 ClpP,7UVU |
sEH 对接 | 直接对接 | Vina-GPU 2.1 | 可溶性环氧化物水解酶,4JNC |
Mpro 对接 | 直接对接 | Vina-GPU 2.1 | SARS-CoV-2 主蛋白酶,6W63 |
TBLR1 对接 | 直接对接 | Vina-GPU 2.1 | WD40 结构域,5NAF(无已知配体) |
靶标选择依据(Appendix G):
- • ClpP:抗癌和抗感染的双重潜在靶标,可被 imipridone 类化合物别构激活;
- • Mpro:SARS-CoV-2 病毒复制必需,结构口袋具有四个亚位点,具有挑战性;
- • sEH:炎症和心血管疾病靶标,具有深疏水口袋,是计算方法的良好基准;
- • TBLR1:MeCP2 互作蛋白,与 Rett 综合征相关,目前无已知小分子配体,代表全新靶标探索。
6.2 GPU 加速对接的引入
技术背景:传统 AutoDock Vina 每次对接耗时数十秒,无法支持 GFlowNet 训练循环(每步评估数十至数百个分子)。
解决方案:集成 Vina-GPU 2.1(Tang et al., 2023),基于 QuickVina 2 算法的 GPU 并行实现,将对接速度提升约 10–30 倍,使直接对接训练成为可能。
对接参数:
- • 穷举度(threads):8000;
- • 搜索深度 :启发式公式 ;
- • 结合口袋中心:由参考 PDB 配体原子坐标均值确定;
- • 分子前处理:RDKit 生成初始构象(ETKDG 算法),UFF 力场优化,转换为 pdbqt 格式。
奖励函数:对接分数(负值,更低 = 更强结合)取负数作为奖励:,(所有对接任务统一)。
6.3 基线方法
方法 | 描述 | 是否保证合成性 |
|---|---|---|
GraphGA | 图遗传算法,GuacaMol 实现 | ❌ |
FGFN | 片段 GFlowNet(Bengio et al., 2021) | ❌ |
FGFN+SA | FGFN + SAScore 奖励惩罚项 | 部分 |
SyntheMol | MCTS + Enamine 反应/构建块 | ✅ |
casVAE | 级联 VAE + 贝叶斯优化 | ✅ |
RGFN | 本文方法 | ✅ |
7. 实验结果分析
7.1 奖励分布对比
主要发现:
- • RGFN 的平均奖励低于 GraphGA(不限合成性的 GA 方法,自由度更高);
- • RGFN 的平均奖励优于 SyntheMol 和 casVAE(同为反应空间方法);
- • 与 FGFN 相比,RGFN 在不同任务上表现互有优劣,整体相当;
- • Senolytics 任务是最显著的例外:FGFN 完全无法发现任何高奖励候选物(推测原因是 FGFN 使用的片段库与已知 senolytics 的化学结构不兼容,加之极稀疏的奖励函数),而 RGFN 成功发现了大量候选物;
- • FGFN+SA 整体性能劣于 FGFN,说明在奖励函数中引入合成性惩罚并不能在保证合成性的同时维持优化质量,反而有所损失。
7.2 模式发现能力(Modes)
模式定义:满足以下条件的分子:
- • 奖励值超过任务特定阈值(sEH: 7, Senolytics: 50, DRD2: 0.95, ClpP: 10);
- • 与所有其他已发现模式的 Tanimoto 相似度 < 0.5(化学骨架多样性)。
模式计算使用 Leader 算法。
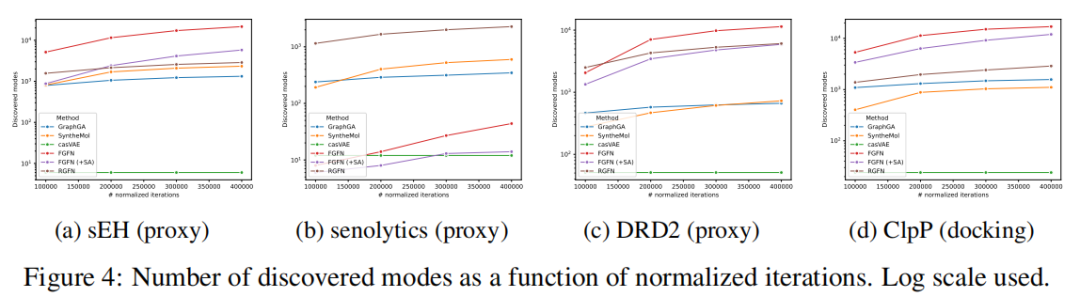
主要发现:
- • 在所有 4 个评测任务中,FGFN 发现的模式数最多(Senolytics 除外),因为其片段空间更灵活,结构多样性更高;
- • RGFN 在所有任务中均优于 SyntheMol、casVAE 和 GraphGA,且在 Senolytics 任务中以绝对优势胜出;
- • RGFN 样本多样性略低于 FGFN,作者认为主要原因是当前使用的片段和反应数目相对有限。
7.3 合成可行性评估
在每种方法生成的 Top-k 模式(Top-500 用于 MW/QED/SAScore,Top-100 用于 AiZynthFinder)上计算以下指标:
指标 | 含义 | 越好 |
|---|---|---|
分子量 (MW) ↓ | 反映分子大小(过大不利于药物化学) | 越小越好 |
QED ↑ | 药物相似性综合评分 | 越高越好 |
SAScore ↓ | 合成可及性启发式评分 | 越低越好 |
AiZynthFinder ↑ | 逆合成规划成功率(0–1) | 越高越好 |
核心数据(部分任务,AiZynthFinder 结果):
任务 | GraphGA | FGFN | FGFN+SA | SyntheMol | casVAE | RGFN |
|---|---|---|---|---|---|---|
sEH | 0.04 | 0.14 | 0.27 | 0.80 | 0.82 | 0.56 |
Senolytics | 0.05 | 0.02 | 0.13 | 0.53 | 0.65 | 0.58 |
ClpP | 0.00 | 0.25 | 0.33 | 0.56 | 0.84 | 0.65 |
DRD2 | 0.41 | 0.76 | 0.78 | 0.66 | 0.87 | 0.87 |
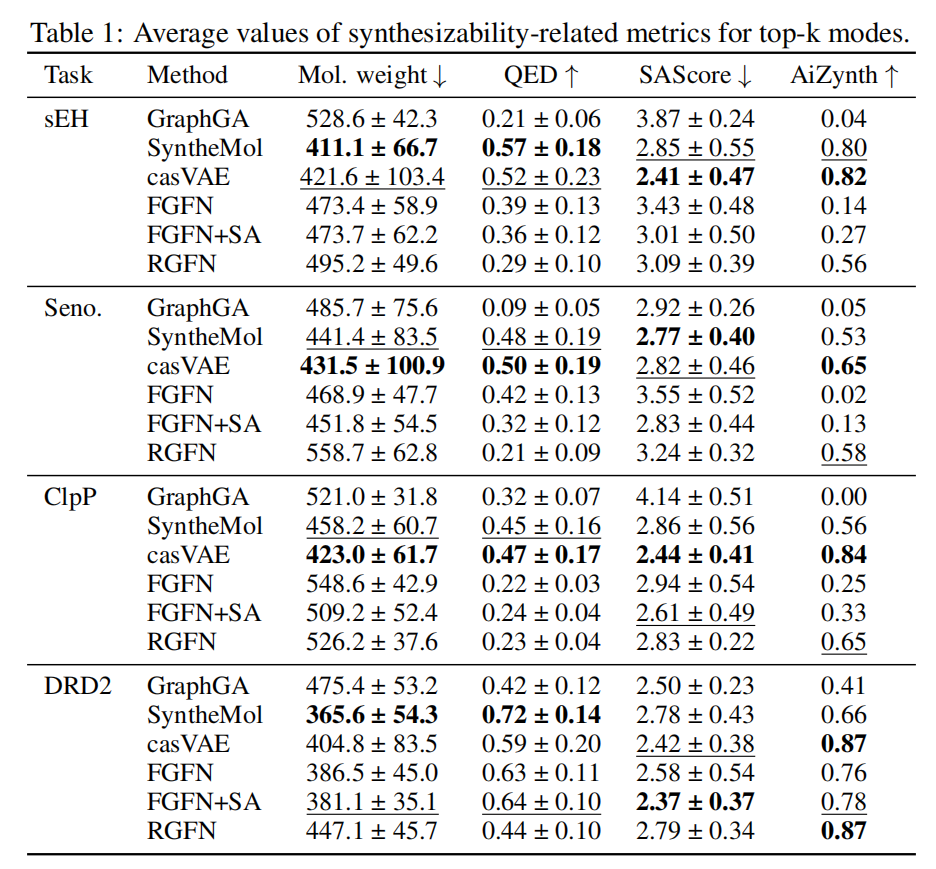
关键解读:
- 1. RGFN 的 AiZynthFinder 成功率显著高于 GraphGA 和 FGFN,接近或达到 SyntheMol/casVAE 水平;
- 2. 即便如此,RGFN 的 AiZynthFinder 评分仍未达到 1.0,但这不代表合成失败——经专业化学家逐一人工审核后,所有 RGFN 模式均被确认为可合成,说明 AiZynthFinder 系统性低估了 RGFN 分子的合成可行性(原因可能是其训练数据与 RGFN 使用的反应类型存在覆盖差距);
- 3. SAScore 虽在形式上是合成性评分,但从 FGFN+SA 案例可以看出,仅优化 SAScore 无法真正保证 AiZynthFinder 层面的合成可行性(AiZynthFinder 仍然很低),说明两种评分之间存在根本性的信息鸿沟。
7.4 片段库规模扩展实验
实验设计:将构建块库从 1k 扩展至 64k,比较独立 index 嵌入与 MACCS 指纹嵌入在训练 2000 步后的骨架发现数量。
结果(以 Murcko 骨架数量衡量):
库规模 | 独立嵌入(median) | 指纹嵌入(median) | 提升倍数 |
|---|---|---|---|
1k | ~90k | ~95k | ~1.05× |
4k | ~82k | ~98k | ~1.2× |
16k | ~72k | ~103k | ~1.4× |
64k | ~60k | ~105k | ~1.75× |
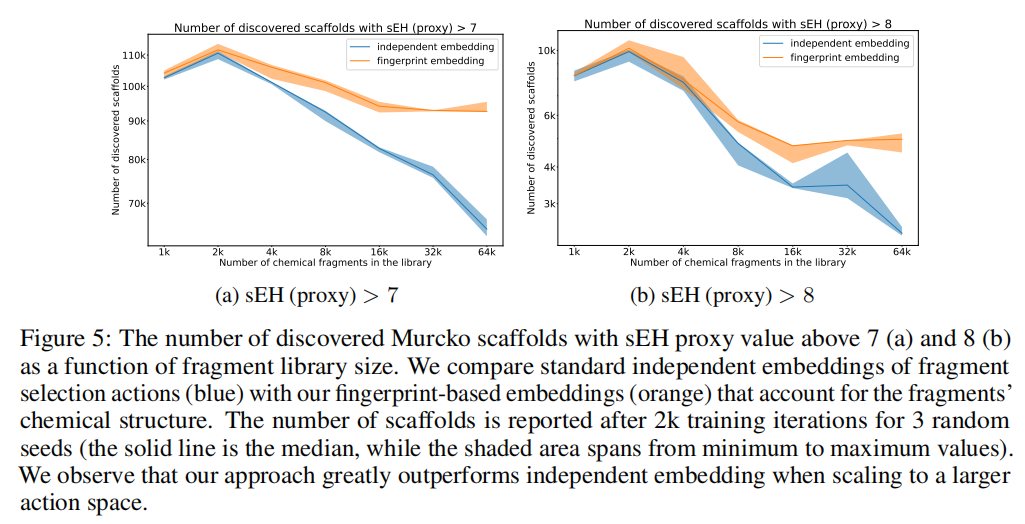
随着库规模增大,指纹嵌入的优势持续扩大。对于 sEH proxy > 8(更严格阈值)的高质量骨架,差异更为显著(在 64k 库下,指纹嵌入约为独立嵌入的 2–3 倍)。
7.5 生成配体的结构多样性与对接姿态
结构多样性(UMAP 分析):对六个任务各取 Top-500 模式的 ECFP 指纹进行 UMAP 降维可视化,结果显示:
- • 不同靶标的生成分子形成明确区分的化学空间簇,证明 RGFN 的化学语言足够丰富,能为不同靶标生成结构独特的候选物;
- • sEH 代理模型任务和 sEH 直接对接任务生成的分子在化学空间中存在一定偏离,提示代理模型对真实对接分数的近似并不完善。
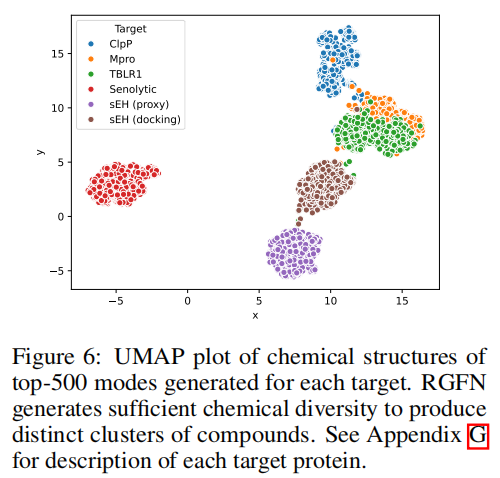
与已知配体的 Tanimoto 相似度:所有任务生成分子与靶标已知 PDB 配体的最大 Tanimoto 相似度中位数约为 0.2–0.3,表明 RGFN 生成的是全新骨架(通常 Tanimoto < 0.4 视为化学骨架不同),而非已知配体的简单类似物。
对接姿态质量(使用PoseBusters 验证):对 Top-100 模式进行 PoseBusters 分子模式检查:
检查项 | sEH | ClpP | Mpro | TBLR1 |
|---|---|---|---|---|
分子加载/清洗 | 100% | 100% | 100% | 100% |
键长合理性 | 100% | 100% | 100% | 100% |
键角合理性 | 100% | 100% | 100% | 100% |
空间冲突 | 100% | 98% | 89% | 96% |
芳环平面性 | 100% | 100% | 100% | 100% |
内部能量 | 90% | 94% | 62% | 34% |
非聚集剂(Tanimoto<0.4) | 38% | 72% | 65% | 53% |
经 PoseBusters 过滤和 Aggregation Advisor 筛选后(排除潜在的非特异性聚集剂),最终保留的高质量模式数分别为:sEH 35、ClpP 68、Mpro 31、TBLR1 15。
TBLR1 内部能量通过率仅 34%:这与该靶标口袋结构特殊(WD40 β 螺旋桨)和目前无已知配体参考有关,是一个值得后续关注的方向。
7.6 成本分析
针对 ClpP 靶标 Top-10 RGFN 配体与 Top-10 SyntheMol 配体进行详细成本核算(仅计原料成本,单位:$/0.1 mmol 产物):
方法 | 平均成本 | 最低 | 最高 |
|---|---|---|---|
RGFN | ~$2.0 | ~$1.4 | ~$3.9 |
SyntheMol | ~$190 | ~$17 | ~$475 |
成本差异原因分析:
- • RGFN 使用的构建块均为大宗化工/医药原料(40/g),通用性强,价格稳定;
- • SyntheMol 使用 Enamine REAL 库的构建块,其中许多是专用的定制合成中间体(624/0.1 g),稀缺性高;
- • 尽管 RGFN 配体需要 4 步反应(55–70% 总收率)vs SyntheMol 约 1 步(90–95% 收率),但原料成本差距(约 100 倍)远超步骤和收率因素。
8. 局限性深度讨论
8.1 化学语言覆盖的局限
当前 17 类反应对于覆盖完整药物化学空间仍然有限:
- • 缺少环化反应:肽大环化、Diels-Alder、闭环复分解等反应可引入刚性三维骨架,对改善选择性和渗透性至关重要;
- • sp³ 碳比例偏低(Fsp³):现有反应倾向于生成平面芳香性化合物,而 Fsp³ 是现代药物化学的重要指标(高 Fsp³ 与溶解度、选择性正相关);
- • 立体化学缺失:SMARTS 模板未编码立体选择性,生成分子无手性中心控制。
8.2 合成路线的不完整性
RGFN 提供的是构建块序列和反应类型序列,严格意义上不等同于完整的合成方案(full synthetic protocol),后者还需要:
- • 具体催化剂和配体选择(如 Pd/Xphos vs Pd/BINAP);
- • 溶剂和温度条件;
- • 保护基策略;
- • 中间体纯化方案。
这些细节对于实际合成成功率有重要影响,需要化学家在 RGFN 建议的路线基础上进一步精细化。
8.3 对接评分函数的内在缺陷
论文坦承分子对接作为 Oracle 的核心局限:
- 1. 分子量偏差:对接分数与分子量强相关(大分子更易获得高分),RGFN 倾向于生成接近 500–550 Da 的较大分子,QED 评分偏低(0.2–0.3);
- 2. 预测精度有限:已知配体的对接分数与 RGFN Top 模式相比并无显著优势(见 Table 4),说明高对接分数并不一定对应高实验活性;
- 3. 口袋特异性差异:不同蛋白口袋对对接方法的适用性差异很大;
近期解决思路:多保真度框架(Multi-fidelity)——在低计算成本对接筛选的基础上,对少数候选物使用 MM-PBSA 或 FEP 进行更精确的结合自由能计算,结合 GFlowNet 的主动学习循环。
8.4 多样性受限
与不约束合成性的 FGFN 相比,RGFN 发现的独特化学骨架(Murcko scaffolds)数量更少。根本原因是:
- • 有限的片段库(350)约束了化学骨架多样性的上限;
- • 反应的兼容性要求(只有特定功能团组合才能参与特定反应)进一步限制了组合多样性。
扩展片段库和增加更多反应类型是解决此问题的直接途径,且论文已通过扩展实验验证了技术可行性。
9. 对药物发现领域的启示
9.1 计算设计与实验验证的鸿沟
当前计算药物发现领域最大的系统性问题之一,是"计算层面高分"与"实验层面可验证"之间的割裂。RGFN 代表了一种思路:不是在生成后试图弥合这一鸿沟,而是在生成过程中直接消除它。
9.2 主动学习闭环的基础设施价值
RGFN 的真正潜力可能在于作为实验-计算主动学习(Active Learning)循环中的核心生成引擎:
初始对接筛选 (RGFN)
↓
合成 Top-N 候选物(~$1–10/化合物)
↓
实验生物活性测试
↓
以实验数据更新 Oracle(代理模型或多保真度模型)
↓
新一轮 RGFN 生成 → 循环低合成成本(100+ 量级)使得每轮闭环可以验证更多候选物,从而更快收敛到真正有活性的化学结构。
9.3 稀有靶标探索的价值
对于 TBLR1 这类目前没有任何已知小分子配体的靶标,RGFN 仅凭结构信息(对接口袋)即生成了 15 个通过所有质量过滤的候选物。这对"孤儿靶标"(orphan target)的药物发现具有直接应用价值。
9.4 化学空间探索的战略意义
当前主流筛选库(包括 DNA 编码化学库 DEL、Enamine REAL 等)在化学空间中的覆盖实际上高度聚集于少数成熟反应和骨架。RGFN 通过精心设计的化学语言,有潜力探索这些库覆盖之外的全新化学空间,为发现具有新型作用机制的候选药物提供可能。
总结:RGFN 是一个在工程实用性与方法创新之间取得良好平衡的工作。它并非追求各项指标的极致(在若干传统基准上仍有差距),而是专注解决一个真实且重要的问题:如何让 AI 生成的药物候选分子真正走出计算机、进入实验室。在这一维度上,RGFN 提供了一个完整、开放、经过验证的技术路线,值得药物发现领域的研究者和工业界团队认真参考与扩展。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-23,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号