ICLR | RxnFlow:面向可合成的AI药物分子生成

ICLR | RxnFlow:面向可合成的AI药物分子生成

DrugIntel
发布于 2026-03-30 16:12:56
发布于 2026-03-30 16:12:56

论文全称:Generative Flows on Synthetic Pathway for Drug Design 发表会议:ICLR 2025 作者机构:KAIST · Simon Fraser University · POSTECH · HITS 通讯作者:Seonghwan Seo、Woo Youn Kim 开源代码:https://github.com/SeonghwanSeo/RxnFlow
目录
- 1. 研究背景与动机
- 2. 相关工作梳理
- 3. 方法框架详解
- • 3.1 GFlowNet 基础回顾
- • 3.2 合成路径动作空间的形式化
- • 3.3 动作空间子采样:核心技术创新
- • 3.4 非层次化 MDP:适配动态库的关键
- • 3.5 模型架构
- 4. 实验设计与结果分析
- • 4.1 口袋特异性优化(Pocket-specific Optimization)
- • 4.2 零样本口袋条件生成(Zero-shot Pocket-conditional Generation)
- • 4.3 无需重训练引入新目标
- • 4.4 动作空间动态扩展
- • 4.5 消融研究
- 5. 理论分析
- 6. 方法局限与未来方向
1. 研究背景与动机
1.1 结构导向药物发现的现状
结构导向药物发现(Structure-Based Drug Discovery, SBDD)是现代药物研发的核心范式之一。随着 AlphaFold 等蛋白质结构预测工具的普及和高分辨率晶体学技术的成熟,研究者可以更便捷地获取靶标蛋白的三维结构,为基于结构的分子设计提供了前所未有的基础。
传统的虚拟筛选方法(Virtual Screening)虽然有效,但面临巨大的计算代价:化学空间估计包含约 10^60 个类药分子,暴力穷举式筛选在计算上根本不可行。这促使研究者转向深度生成模型,期望通过学习分子-靶标相互作用的分布来直接生成候选分子。
1.2 合成可及性:被忽视的核心瓶颈
现有深度生成模型(包括扩散模型、VAE、GAN 等)在提升分子对接分数和药物类药性方面已取得显著进展,但普遍存在一个致命弱点:生成的分子往往无法在实验室中被合成。
这一问题的严重性体现在多个层面:
- • 合成可及性评估工具的局限:常用的合成可及性评分(SA Score,Ertl & Schuffenhauer, 2009)基于片段频率统计,本质上是一种启发式近似,无法反映真实的逆合成路径可行性。研究表明,SA Score 与实际合成复杂度的相关性相当有限。
- • 后验投影方法的代价:另一类方法尝试将生成模型输出的分子"投影"到可合成化学空间(如 Gao et al., 2022b;Luo et al., 2024),但这一过程不可避免地改变分子结构,可能降低已优化的生物活性。
- • 湿实验验证的现实门槛:在工业药物研发流程中,一个分子若无法以合理的步骤数、成本和产率被合成,则其计算层面的优越性毫无实用价值。
1.3 合成路径生成:一条更根本的出路
一种从根本上解决合成可及性问题的思路是:将合成路径直接内嵌进分子生成过程。具体而言,将分子生成建模为一个顺序决策过程:从商业可购买的砌块(Building Blocks)出发,按照真实的化学反应模板逐步组装,每一个生成的分子自动附带其合成路径。
这一思路的工业背景是 Enamine REAL 等虚拟库的出现——通过组合枚举商业砌块和反应模板,可以构建出数十亿乃至上百亿规模的"按需可合成"化学空间,其中每个分子都有明确的合成方案。
然而,这条路线也面临一个核心挑战:动作空间规模巨大。当砌块库包含百万级分子时,每一步反应的候选动作空间极其庞大,导致传统方法不得不大幅压缩可用砌块数量,牺牲化学多样性换取计算可行性。
RxnFlow 的核心贡献,正是在不妥协化学多样性的前提下,解决这一规模瓶颈。
2. 相关工作梳理
2.1 基于结构的深度生成模型
类型 | 代表方法 | 原理 | 局限 |
|---|---|---|---|
原子级生成 | TargetDiff, DiffSBDD, DecompDiff | 扩散模型直接生成原子坐标 | 合成可及性差 |
自回归模型 | Pocket2Mol | 逐原子/片段自回归 | 合成可及性差 |
贝叶斯流网络 | MolCRAFT | 连续参数空间生成 | 合成可及性差 |
GFlowNet | TacoGFN | 片段组装 + 奖励比例采样 | SA Score 近似不精准 |
2.2 合成导向生成模型
方法 | 核心思想 | 局限 |
|---|---|---|
Bradshaw et al. (2019) | 合成路径建模为 DAG | 规模受限 |
Horwood & Noutahi (2020) | MDP + 强化学习 | 仅优化单一目标 |
SynNet (Gao et al., 2022b) | 遗传算法优化合成树 | 多样性不足 |
BBAR (Seo et al., 2023) | 条件生成模型 | 无法进行在线优化 |
SynFlowNet (Cretu et al., 2024) | 反应基 GFlowNet | 砌块库规模受限(6k-220k),层次化 MDP 适应性差 |
RGFN (Koziarski et al., 2024) | 反应基 GFlowNet | 砌块库规模受限(350-64k),层次化 MDP 适应性差 |
2.3 GFlowNet 框架
GFlowNet(Bengio et al., 2021)是一种训练随机生成策略的学习框架,使生成对象的概率正比于其奖励函数值。与仅最大化奖励的强化学习不同,GFlowNet 天然支持从多个高奖励模式中采样,这对于需要探索化学多样性的药物发现任务尤为重要。
训练目标方面,本文采用 轨迹平衡(Trajectory Balance, TB)目标(Malkin et al., 2022),其相比流匹配(Flow Matching)和详细平衡(Detailed Balance)具有更好的信用分配特性和收敛稳定性。
3. 方法框架详解
3.1 GFlowNet 基础回顾
GFlowNet 在有向无环图 上定义生成过程,其中 为可达状态空间, 为动作空间。给定终止状态 ,轨迹流定义为 ,即终止状态的奖励值。
流匹配条件(中间状态):
边界条件:
- • 终止状态:
- • 配分函数:
前向/后向策略:
满足上述条件时,生成策略自动实现 。
轨迹平衡损失(本文训练目标):
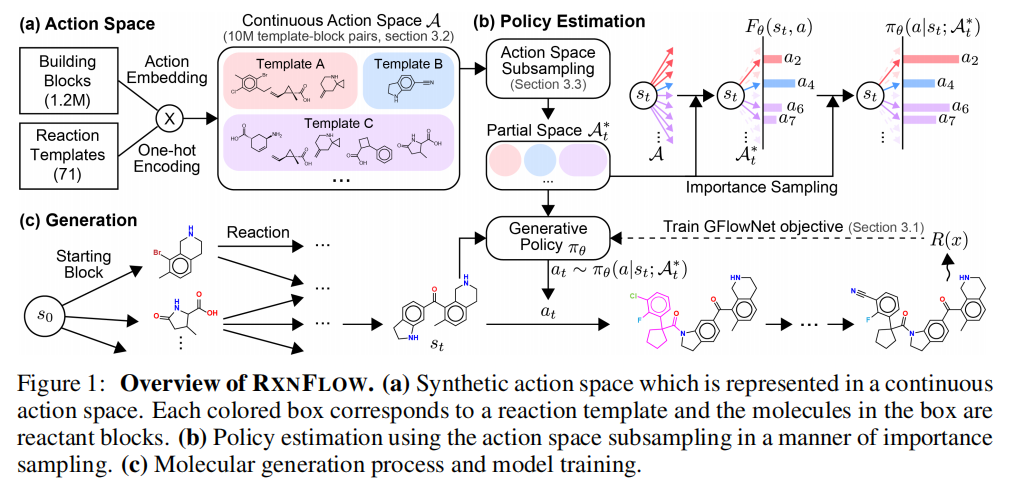
3.2 合成路径动作空间的形式化
本文遵循 Cretu et al. (2024) 的框架,将化学反应建模为状态转移,合成路径建模为轨迹。动作空间定义如下:
初始状态 :执行 AddFirstReactant,从砌块集合 (120万个 Enamine 商业砌块)中选择起始分子。
后续状态 :可选动作为:
- •
Stop:终止当前轨迹 - •
ReactUni:执行单分子反应(13种模板) - •
ReactBi:执行双分子反应,选择模板 (58种,考虑反应物顺序后为116种)及对应砌块
形式化为:
其中,与当前分子不相容的反应模板会被掩码(masking)。整个动作空间规模约为 数量级(模板-砌块对),这是导致计算瓶颈的直接原因。
3.3 动作空间子采样:核心技术创新
3.3.1 问题定义
对于后续状态,双分子反应的动作数量级为 ,在 GPU 显存和计算时间上均不可承受。SynFlowNet 和 RGFN 通过限制砌块库大小(6k-220k)来规避这一问题,代价是丧失化学多样性。
3.3.2 子采样策略
RxnFlow 引入辅助子采样策略,在每步转移时从完整动作空间 中采样一个子集 :
- • 初始状态:从 均匀采样子集 (采样比例 1%)
- • 后续状态:保留全部
Stop和ReactUni动作(数量少,不能丢弃稀有动作类型);对每个双分子模板 ,均匀采样子集
计算复杂度从 降至 。
3.3.3 重要性采样估计前向策略
为从子集 无偏地估计状态流 ,对每个动作赋予权重:
估计状态流:
可以严格证明,该估计是无偏的:
3.3.4 偏差分析
子采样引入的轨迹平衡损失偏差可以量化为:
关键结论:偏差与采样数量()强相关,与采样比例()弱相关。这意味着只要保证足够的绝对采样量,即使面对百万级砌块库,偏差也可控。玩具实验验证了在 1% 采样比下, 的偏差小于 0.005,轨迹平衡损失与真实值几乎一致。
3.4 非层次化 MDP:适配动态库的关键
3.4.1 层次化 MDP 的固有缺陷
SynFlowNet 和 RGFN 采用层次化 MDP:先选择反应模板 ,再从 中选择砌块 :
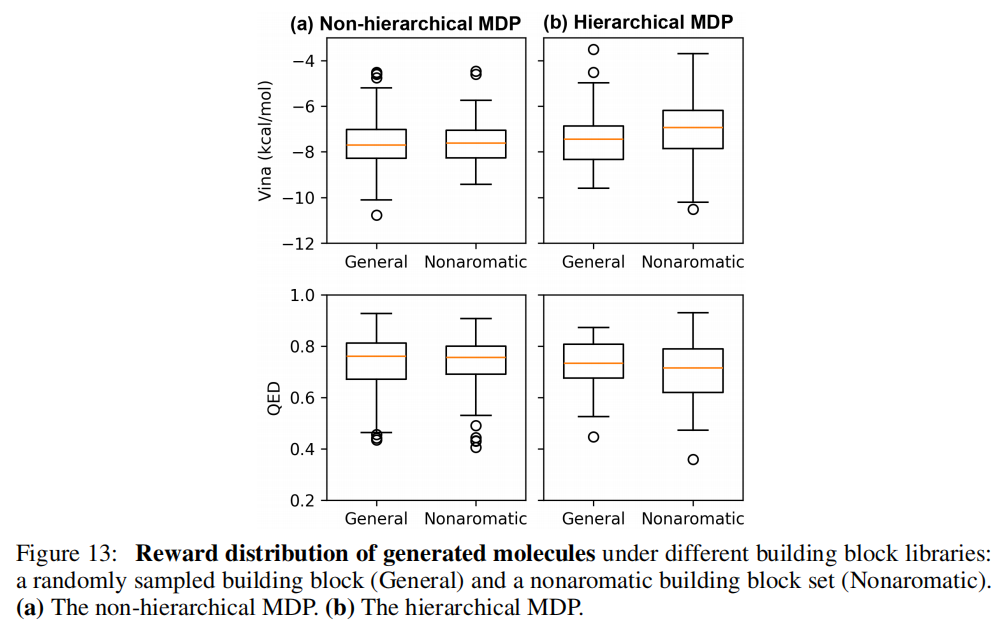
问题在于:模板选择概率在训练后固定。若修改砌块库(如排除含有特定官能团的砌块),模板的流量估计会出现系统性偏差——某些模板下可用砌块大幅减少,但模板本身的选择概率却无法相应调整(如图 2(a) 所示)。
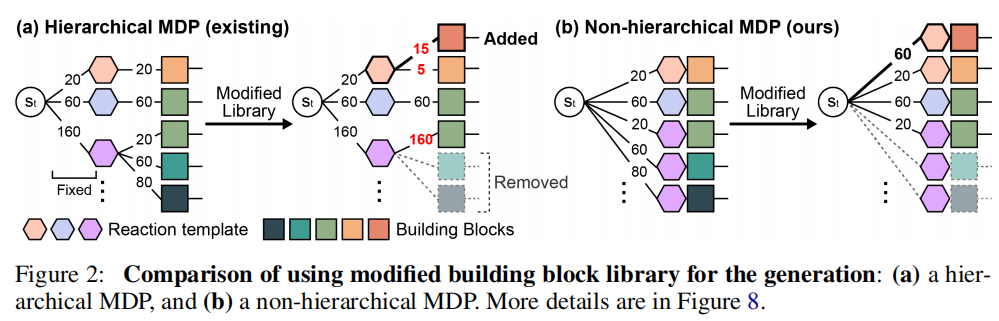
3.4.2 非层次化 MDP 的设计
RxnFlow 联合选择(模板, 砌块)对:
当砌块库发生变化时,分母自动重新归一化,无需重训练即可获得准确的策略估计。这一特性使 RxnFlow 能够:
- 1. 动态排除砌块:无需重训练引入结构约束(如毒性过滤)
- 2. 整合新砌块:利用动作嵌入泛化到未见砌块
- 3. 调整优化目标:仅通过修改可用砌块集合实现新目标
当然,非层次化 MDP 的代价是计算开销更大(分母需要遍历所有模板-砌块对),但动作空间子采样恰好弥补了这一不足。
3.5 模型架构
3.5.1 动作嵌入(Action Embedding)
为避免为每个砌块维护独立参数(这在百万级砌块下不可行),RxnFlow 使用一个共享网络 将砌块 编码为连续向量,输入特征包括:
- • 理化性质(8维):分子量、原子数、HBA/HBD、芳香/非芳香环数、LogP、TPSA
- • MACCS 指纹:反映官能团组成
- • Morgan ECFP4 指纹(1024维):反映拓扑结构信息
边流计算(以双分子反应为例):
其中 为反应模板的 one-hot 编码, 表示向量拼接。
3.5.2 主干网络
- • 分子状态编码:图变换器(Graph Transformer,Yun et al., 2022)
- • 口袋条件生成:GVP-GNN 编码蛋白口袋残基图(K 近邻图)
- • 3D 交互建模(实验性扩展):利用对接构象构建配体-蛋白 3D 复合图
3.5.3 后向策略
不同于简单均匀后向策略,本文设计的后向转移概率正比于沿该入边到达当前状态的期望轨迹数量,以鼓励较短的合成路径(对应于更低的合成复杂度和成本)。
4. 实验设计与结果分析
4.1 口袋特异性优化
实验设置
- • 数据集:LIT-PCBA 中的 15 个蛋白靶标
- • 对接工具:GPU 加速的 UniDock(Vina 评分函数),在线训练期间每步生成 64,000 个分子
- • 奖励函数:,采用多目标 GFlowNet(Jain et al., 2023)框架
- • 质量约束:QED > 0.5,Tanimoto 距离 > 0.5(保证结构多样性),每个靶标选 Top-100 候选
- • 合成可及性验证:使用 AiZynthFinder 进行逆合成分析,仅以 USPTO 反应和 Enamine 砌块为依据
核心结果
命中率(Hit Ratio):分子同时满足合成可及性(AiZynthFinder 验证)且对接分优于已知参考配体的比例
方法 | ADRB2 | ALDH1 | ESR_ago | ESR_antago | FEN1 |
|---|---|---|---|---|---|
FragGFN | 4.00 | 3.75 | 0.25 | 0.25 | 0.25 |
SynFlowNet | 52.75 | 57.00 | 30.75 | 11.25 | 53.00 |
RGFN | 46.75 | 39.75 | 4.50 | 1.25 | 19.75 |
RxnFlow | 60.25 | 63.25 | 71.25 | 46.00 | 65.50 |
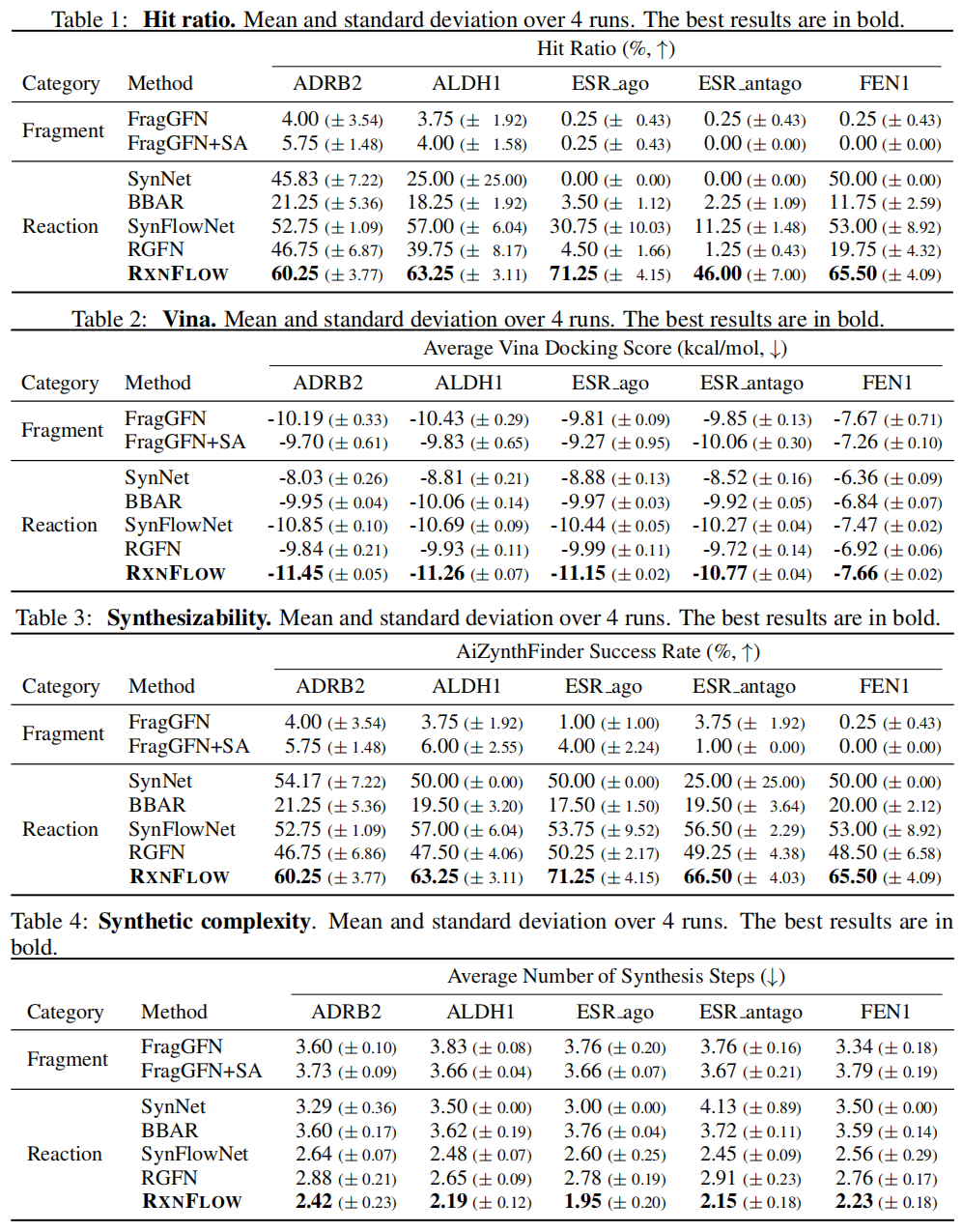
合成步骤数(越少越好,关联合成成本和产率):
方法 | 平均步骤数 |
|---|---|
FragGFN | ~3.7 |
SynFlowNet | ~2.55 |
RGFN | ~2.80 |
RxnFlow | ~2.15 |
关键发现:RxnFlow 在使用更多砌块(120万 vs SynFlowNet 的 6千)的同时,反应步骤数却更少(平均约 2.15 步 vs SynFlowNet 约 2.55 步)。这验证了核心论点:更大的砌块库允许以较少步骤组装出同等复杂的分子,从而降低整体合成复杂度。
同时,RxnFlow 甚至超越了不施加合成约束的 FragGFN,表明 Enamine 砌块库作为类药化学空间的天然约束,以及较短轨迹对轨迹平衡目标的优化优势,共同带来了性能提升。
4.2 零样本口袋条件生成
实验设置
- • 基准数据集:CrossDocked2020(100个测试口袋,每个生成100个分子)
- • 评价指标:Vina 对接分(QuickVina)、QED、合成可及性(AiZynthFinder)、Tanimoto 多样性
- • 对照策略:使用基于药效团表示的对接代理模型(PharmacoNet 代理),无需对每个靶标重新训练
CrossDocked2020 基准结果
方法 | Vina (avg) | Vina (med) | QED (avg) | 合成可及率 | 多样性 | 生成时间(s) |
|---|---|---|---|---|---|---|
参考配体 | -7.71 | -7.80 | 0.48 | 36.1% | — | — |
Pocket2Mol | -7.60 | -7.16 | 0.57 | 29.1% | 0.83 | 2504 |
TargetDiff | -7.37 | -7.56 | 0.49 | 9.9% | 0.87 | 3428 |
DecompDiff | -8.35 | -8.25 | 0.37 | 0.9% | 0.84 | 6189 |
MolCRAFT | -8.05 | -8.05 | 0.50 | 16.5% | 0.84 | 141 |
MolCRAFT-large | -9.25 | -9.24 | 0.45 | 3.9% | 0.82 | >141 |
TacoGFN | -8.24 | -8.44 | 0.67 | 1.3% | 0.67 | 4 |
RxnFlow | -8.85 | -9.03 | 0.67 | 34.8% | 0.81 | 4 |
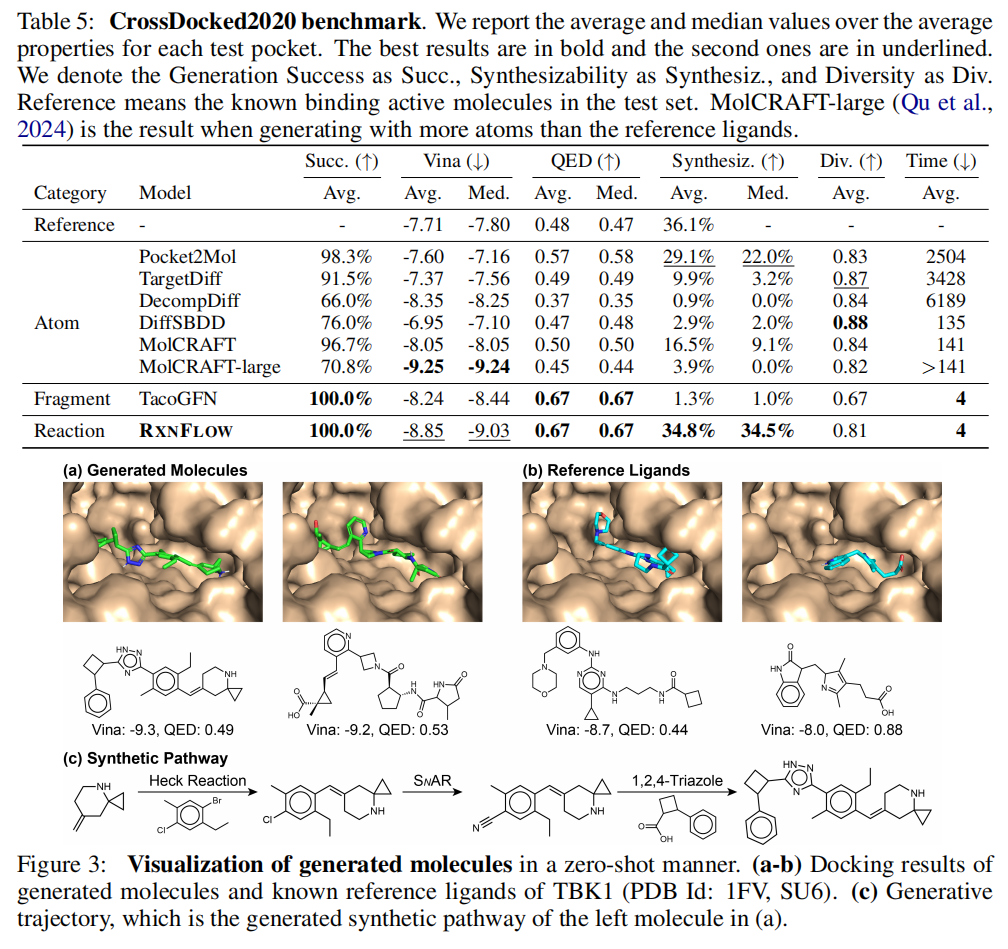
深度解读
这一结果揭示了一个极为重要的现象:RxnFlow 是唯一在活性、药物性和合成可及性三个维度上同时达到实用水准的方法。
- • MolCRAFT-large 对接分最优(-9.25),但合成可及率仅 3.9%——几乎所有分子都无法合成
- • DecompDiff 对接分次优(-8.35),合成可及率更是跌至 0.9%
- • Pocket2Mol 合成可及率较好(29.1%),但对接分(-7.60)甚至劣于参考配体
- • RxnFlow 的合成可及率(34.8%)不仅远超其他模型,更超过了参考活性配体(36.1%)——这意味着 RxnFlow 生成的分子在合成便利性上与已知药物分子相当
多样性方面,RxnFlow(0.81)显著优于 TacoGFN(0.67),接近基于分布学习的模型(0.83-0.87),这一优势来源于 Enamine 砌块库的化学多样性远超片段集合。
靶标特异性(Delta Score):RxnFlow 在靶标特异性上与 TacoGFN 持平(-1.13),略低于 MolCRAFT(-2.08),表明其生成的分子对目标口袋具有合理的特异性,但仍有提升空间。
4.3 无需重训练引入新目标
以在 Sec 4.2 预训练模型上额外引入溶解性目标为例:针对 KRAS-G12C 靶标,通过将 AddFirstReactant 和 ReactBi 的可用砌块限制为 TPSA 后 15% 的低极性砌块,无需任何模型参数更新,生成的分子 TPSA 分布显著下移(更疏水),而总体奖励分布(Vina + QED)几乎不变。
这一能力依赖于非层次化 MDP 的设计:排除部分砌块后,策略估计自动重新归一化,无需重训练。相比之下,层次化 MDP 方法在修改砌块库后会出现策略估计偏差(图 13 对比结果)。
4.4 动作空间动态扩展
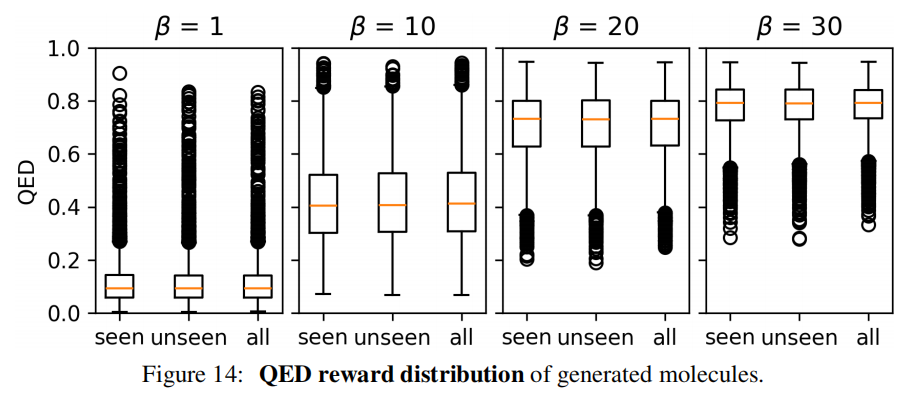
将 100 万砌块分为 50 万"已见"和 50 万"未见"两部分,仅用"已见"砌块训练。实验结果显示,在多个奖励指数()设置下,使用"未见"砌块生成的分子奖励分布与"已见"砌块几乎完全一致,证明动作嵌入实现了对化学结构的有效泛化。
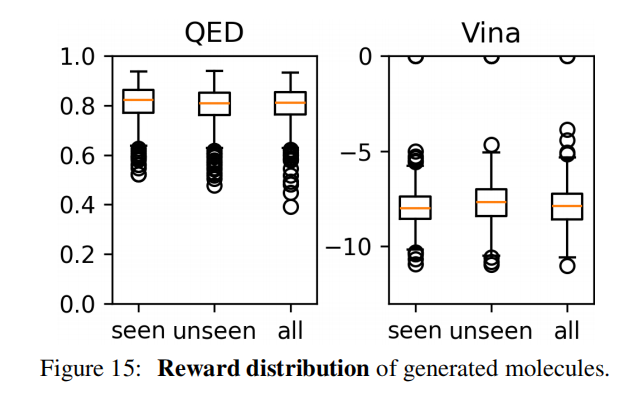
进一步实验(图 15)中,将"未见"砌块限制为与训练砌块 Tanimoto 相似度 < 0.5 的结构差异较大的砌块,结果同样稳定,验证了模型对分布外砌块的鲁棒性。
4.5 消融研究
砌块库大小的影响(图 6,OPRK1 靶标):
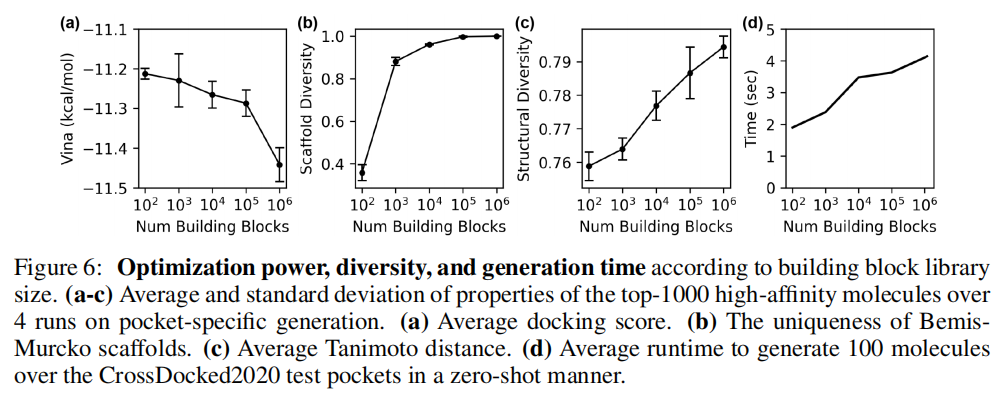
随着砌块库从 100 个扩展至 100 万个:
- • 平均对接分单调提升(从 -11.1 到 -11.5 kcal/mol)
- • Bemis-Murcko 骨架唯一性持续增加
- • Tanimoto 多样性持续增加
- • 生成时间:砌块库扩大 100 倍,生成时间仅增加约 2 倍——体现了动作空间子采样的计算效率优势
3D 交互建模(探索性实验):使用对接构象构建 3D 复合图作为状态输入,相比 2D 图有显著提升(图 7),验证了引入 3D 空间信息的潜力,但该功能目前仍处于探索阶段,因计算开销较大未纳入主实验。
5. 理论分析
5.1 无偏估计的严格证明
对于均匀子采样策略,设 为大小 的集合, 为大小 的均匀子集,则估计量 的方差为:
对应 的方差(通过 Delta 方法近似):
这说明偏差随采样数量 线性可控,当 时方差趋于零。
5.2 后向策略的设计
在合成路径 DAG 中,每个状态的出边远多于入边(向前探索的可能性远多于反应物来源),且从初始状态到某状态的轨迹数量随距离增加而指数增长。均匀后向策略会导致长路径轨迹的流量被系统性低估,从而偏向短路径。RxnFlow 的后向策略设计正比于期望轨迹数量,纠正了这一偏差,有效支持不同长度的合成路径。
6. 方法局限与未来方向
6.1 当前局限
- 1. 子采样引入的训练偏差:虽然理论上偏差可控,且实验表明在 1% 采样率下偏差较小,但在极端情况下(如某些双分子反应的可用砌块数量极少)仍需特殊处理(论文设置最小采样量为 100)。
- 2. 代理模型依赖:口袋条件生成中使用的对接代理模型(PharmacoNet)引入了近似误差,对接分可能与实际物理结合能存在偏差。
- 3. 反应模板的局限:使用的 71 种反应模板来自公开数据集,可能无法覆盖所有合成可行路线;同时,模拟计算结果与实际产率可能存在差距。
- 4. 3D 信息未充分利用:主模型仍基于 2D 分子图状态,3D 对接构象信息仅在探索性实验中使用,计算代价较高,尚未规模化应用。
- 5. 均匀子采样的探索偏差:当前子采样策略均匀随机选取砌块,可能对优质砌块的利用不够充分。
6.2 未来方向
论文作者指出两个明确的改进方向:
- 1. 引入 3D 交互建模:在不显著增加计算成本的前提下,将对接构象信息纳入状态表示,改善生成决策的物理合理性(已有初步实验验证可行性)。
- 2. 优先子采样:将当前均匀子采样策略升级为基于砌块质量评分的优先采样,在保证计算效率的同时提高高质量砌块的利用率,更好平衡探索(exploration)和利用(exploitation)。
此外,潜在的扩展方向还包括:多步逆合成约束(确保合成路径在每一步都满足实际合成条件)、与实验数据的主动学习结合、以及对非 Enamine 砌块库的适配。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-28,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号