PNAS | AlphaFold3 的「记忆偏见」:它真的在预测配体诱导的构象变化,还是在回忆训练数据?

PNAS | AlphaFold3 的「记忆偏见」:它真的在预测配体诱导的构象变化,还是在回忆训练数据?

DrugIntel
发布于 2026-03-30 16:13:20
发布于 2026-03-30 16:13:20

论文精读 | PNAS 2026, Vol. 123, No. 10 原文标题:Bias in the AlphaFold3 prediction of ligand-induced domain motion in enzymes 作者:Hao Yu, Ayse A. Bekar-Cesaretli, Maria Lazou, Dima Kozakov, Diane Joseph-McCarthy, Sandor Vajda 单位:Boston University; The University of Texas at Austin DOI:10.1073/pnas.2530709123 | 发表日期:2026年3月4日
一、为什么要读这篇论文?
自2021年 AlphaFold2 横空出世以来,深度学习驱动的蛋白质结构预测已经深刻改变了结构生物学的面貌。2024年,AlphaFold3(AF3)进一步将预测能力扩展到蛋白质-配体、蛋白质-核酸等多分子复合物的"共折叠"(cofolding)。一个自然的期望是:既然 AF3 可以同时接受蛋白质序列和配体 SMILES 作为输入,它应该能够建模配体诱导的蛋白质构象变化——这对药物设计和酶催化机制的理解十分重要。
然而,这篇发表于 PNAS 的论文通过系统性的大规模实验,揭示了一个令人不安的事实:AF3 对蛋白质构象的预测,在很大程度上是由训练数据中已有结构的数量比例决定的,而非由配体在计算中的实际存在与否驱动。 这种"记忆化"(memorization)效应不仅存在于 AF3 中,在 AF2 中同样显著。更令人担忧的是,已知不会结合的配体也能诱导类似的构象变化,而 pLDDT 置信度分数不足以可靠地区分真正的结合物与非结合物。
这些发现对于所有使用 AlphaFold 系列工具进行蛋白质构象预测、虚拟筛选和药物设计的研究者来说,都具有重要的参考价值。
二、研究背景
2.1 酶的配体诱导结构域运动
许多酶在行使催化功能时,并非保持静态结构,而是经历显著的构象变化。典型的模式是:酶首先以开放构象(apo/open state)结合底物,随后结构域运动使酶转变为闭合构象(holo/closed state),将底物包裹在高度特异性的催化环境中。这种配体诱导的结构域运动在激酶、异构酶、转移酶等多种酶家族中普遍存在,其幅度可达 10–15 Å RMSD,远超一般的蛋白质主链柔性波动。
DynDom 数据库系统收录了此类酶的信息,每种酶由一对 apo 和 holo X 射线晶体结构代表,并标注了触发结构域运动的"trigger ligand"(触发配体)、固定结构域和运动结构域的残基范围。
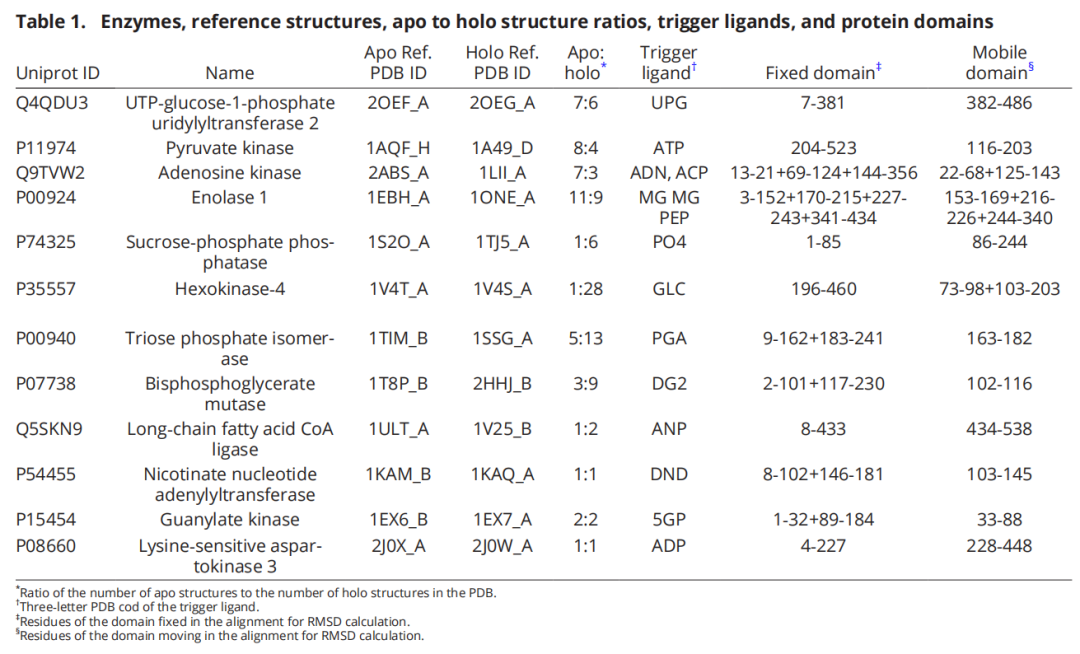
2.2 AF3 的共折叠能力与局限
AF3 的一大进步在于能够处理多分子输入,包括蛋白质序列和配体 SMILES 字符串。理论上,通过分别运行"仅蛋白质序列"和"蛋白质序列 + 配体 SMILES"两组计算,可以比较 AF3 预测的 apo 和 holo 构象,评估其建模配体诱导构象变化的能力。
已有研究表明,AF3 在预测相对静态的蛋白质-配体复合物结构方面优于传统对接方法,但对于涉及显著主链重排(>2–5 Å RMSD)的情况仍然是重大挑战。本文所研究的构象变化幅度更大(10–15 Å RMSD),理应更加困难。不过,一个缓解条件是:DynDom 数据库中的大多数结构都在 AF3 的训练集中——而这恰恰成为了问题的根源。
2.3 记忆化问题的已有线索
在本文之前,已有多项研究指出 AF2 和其他 ML 方法存在记忆化问题:
- • Chakravarty 等(2024)发现 AF2 对折叠开关蛋白的预测受结构记忆驱动
- • Lazou 等(2024)发现 AF2 在预测配体结合位点的多构象时可能"记住太多"
- • Bryant & Noe(2024)和 Schafer & Porter(2025)表明,从训练集中移除结构会改变 AF2 的预测结果
- • Škrinjar 等(2025)和 Masters 等(2025)对蛋白质-配体共折叠方法的记忆化进行了评估
本文在此基础上,首次系统、定量地揭示了 AF3 在酶配体诱导结构域运动建模中的记忆偏见。
三、实验设计
3.1 蛋白质数据集
从 DynDom 数据库中选取 82种具有配体诱导结构域运动的酶,每种酶拥有明确的 apo 和 holo 参考结构,以及已知的触发配体。
3.2 模型生成
对每种酶,使用 MMseqs2(Release 18)生成多序列比对(MSA)。AF3 使用默认参数,不使用模板结构:
- • 无配体组:输入 = 蛋白质序列 + MSA → 100个随机种子 × 5个预测 = 500个模型
- • 有配体组:输入 = 蛋白质序列 + MSA + 配体 SMILES → 100个随机种子 × 5个预测 = 500个模型
3.3 评估方法
所有模型和 PDB 结构在二维坐标系中展示:X 轴为与 apo 参考结构的 RMSD,Y 轴为与 holo 参考结构的 RMSD(对齐固定结构域后计算运动结构域的 RMSD)。每个模型被分类为"更接近 apo"或"更接近 holo"。同时评估配体放置精度(配体 RMSD)和预测置信度(pLDDT)。
3.4 关键分组策略
根据 PDB 中每种酶 apo 与 holo 结构的数量比,将82种酶分为三组:
分组 | 定义 | 蛋白数量 | 含义 |
|---|---|---|---|
Group 1 | apo 结构多于 holo 结构 | 19 | 训练集中开放态占优 |
Group 2 | holo 结构多于 apo 结构 | 25 | 训练集中闭合态占优 |
Group 3 | 总结构 ≤5 或 apo = holo | 38 | 训练数据稀少或均衡 |
这一分组是理解全文结果的核心框架。
四、核心发现
4.1 发现一:预测构象由训练数据比例主导,而非配体存在
这是本文最核心的结论。
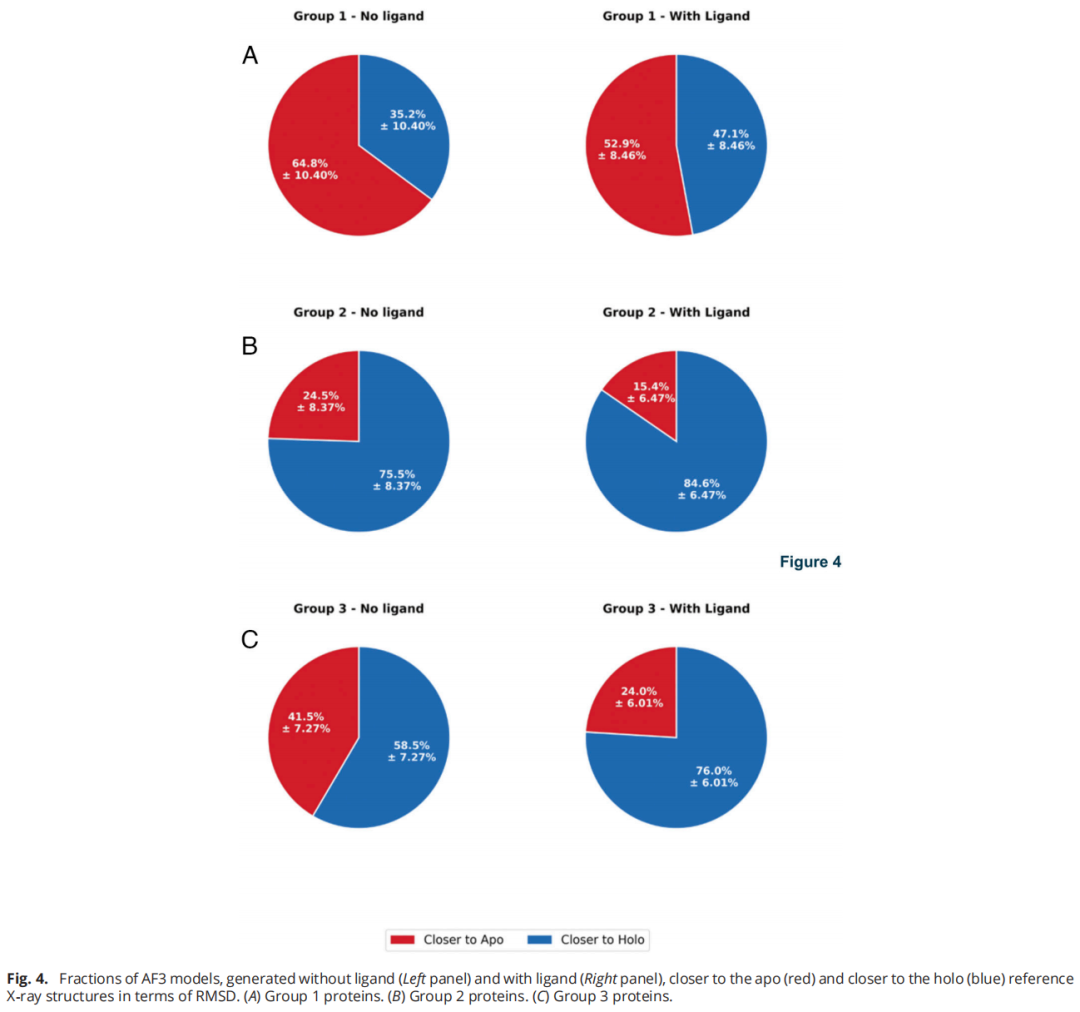
Group 1(apo 占优的酶):无配体时,64.8% 的模型更接近 apo 参考结构。加入配体后,接近 apo 的比例仅降至 52.9%,即配体只让 11.9% 的模型从 apo 侧转向 holo 侧。
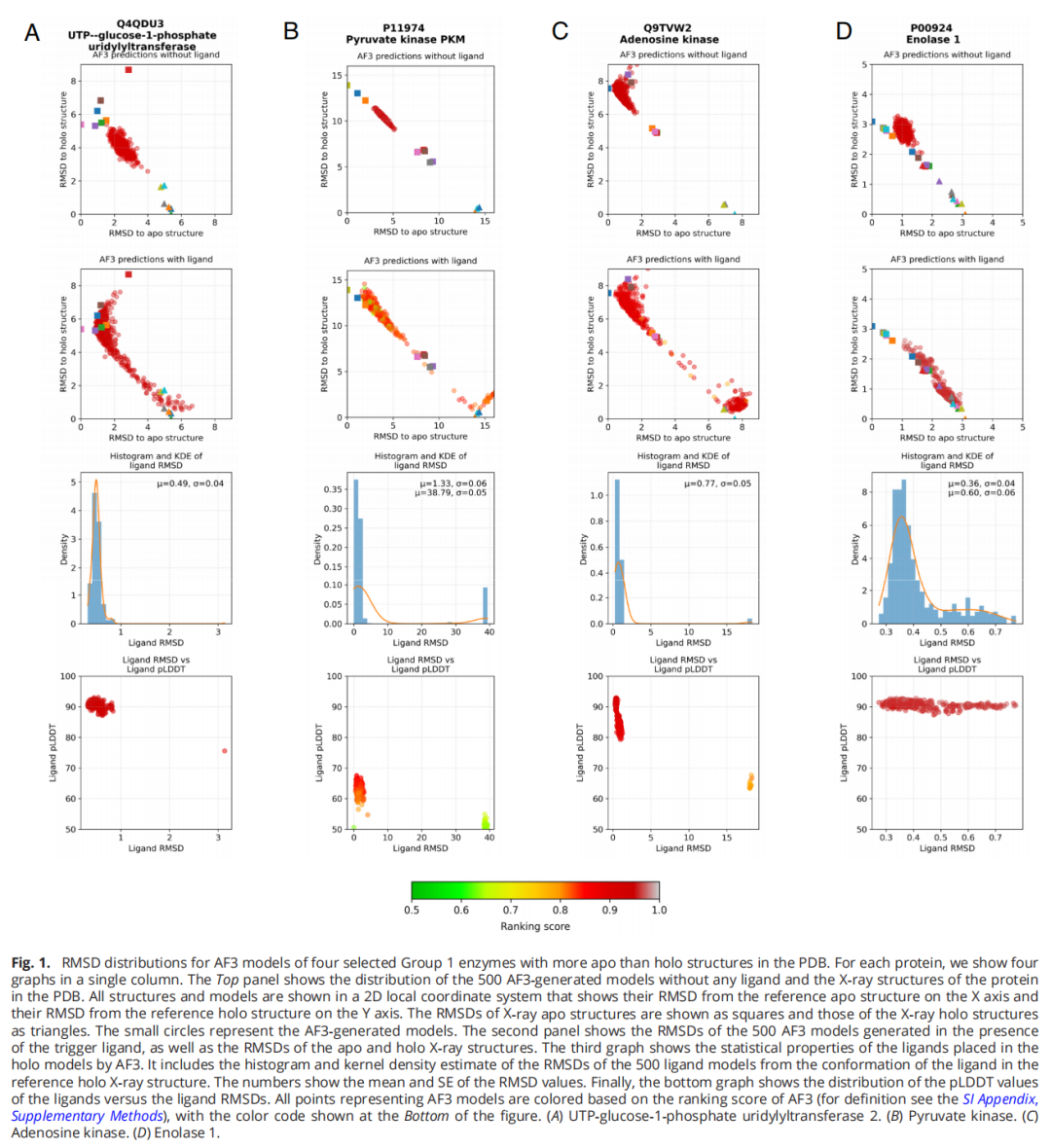
Group 2(holo 占优的酶):无配体时,75.5% 的模型已经更接近 holo 参考结构——尽管计算中根本没有提供任何配体。加入配体后,比例上升至 84.6%,增幅仅 9.1%。
关键对比:
指标 | 数值 | 含义 |
|---|---|---|
Group 1 与 Group 2 holo 模型比例差 | 40.3% | 训练集组成的影响 |
Group 1 加配体后 holo 模型增量 | 11.9% | 配体的实际影响 |
Group 2 加配体后 holo 模型增量 | 9.1% | 配体的实际影响 |
训练数据组成带来的效应(40.3%)是配体实际作用(9–12%)的 3.5–4.5 倍。 这是非常强烈的记忆化证据。
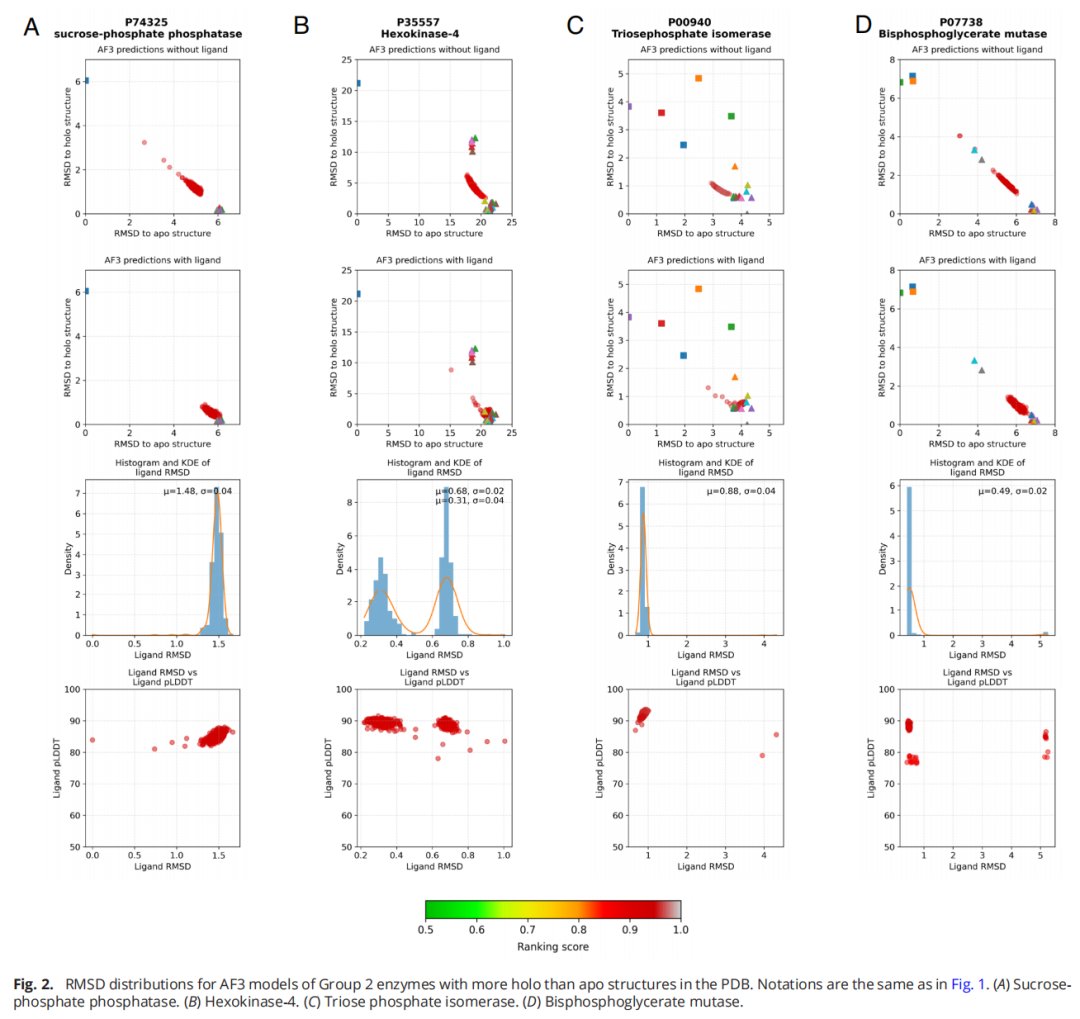
Group 3(数据稀少的酶):配体的影响相对更大,将 holo 模型比例从 58.5% 提升至 76.0%(增加 17.5%)。这说明在记忆偏差较弱的情况下,AF3 确实能更多地依赖配体信息进行预测。
4.2 发现二:配体放置精度与训练数据丰富程度正相关
AF3 在不同组中放置配体的精度差异显著:
分组 | 配体 RMSD < 2Å 的比例 | 平均配体 RMSD | 解释 |
|---|---|---|---|
Group 1 | 42% | 3.57 Å | 结合位点多处于开放态,口袋未形成 |
Group 2 | 80% | 4.61 Å(受离群值影响) | 结合位点默认已闭合,利于配体放置 |
Group 3 | 73.7% | — | 配体促进闭合,口袋形成良好 |
这一结果看似矛盾——Group 2 的配体放置更准确,但这并非 AF3 "理解"了配体结合的物理化学原理,而是因为训练数据中大量 holo 结构使得结合位点默认处于闭合态,配体恰好被放入了一个预先形成好的口袋中。
对于 Group 1 的酶,即使加入配体,仍有 52.9% 的模型停留在开放态,结合位点尚未形成,自然无法准确放置配体。
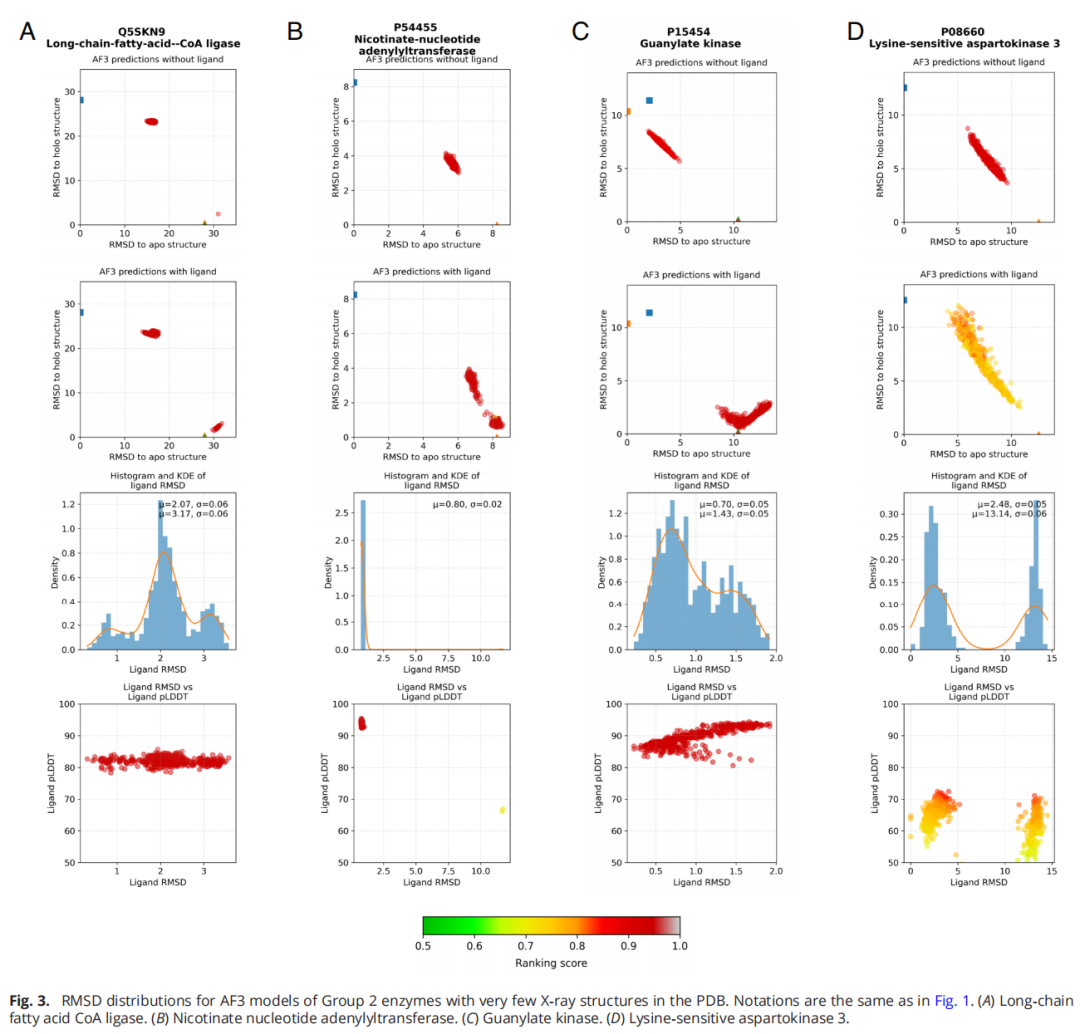
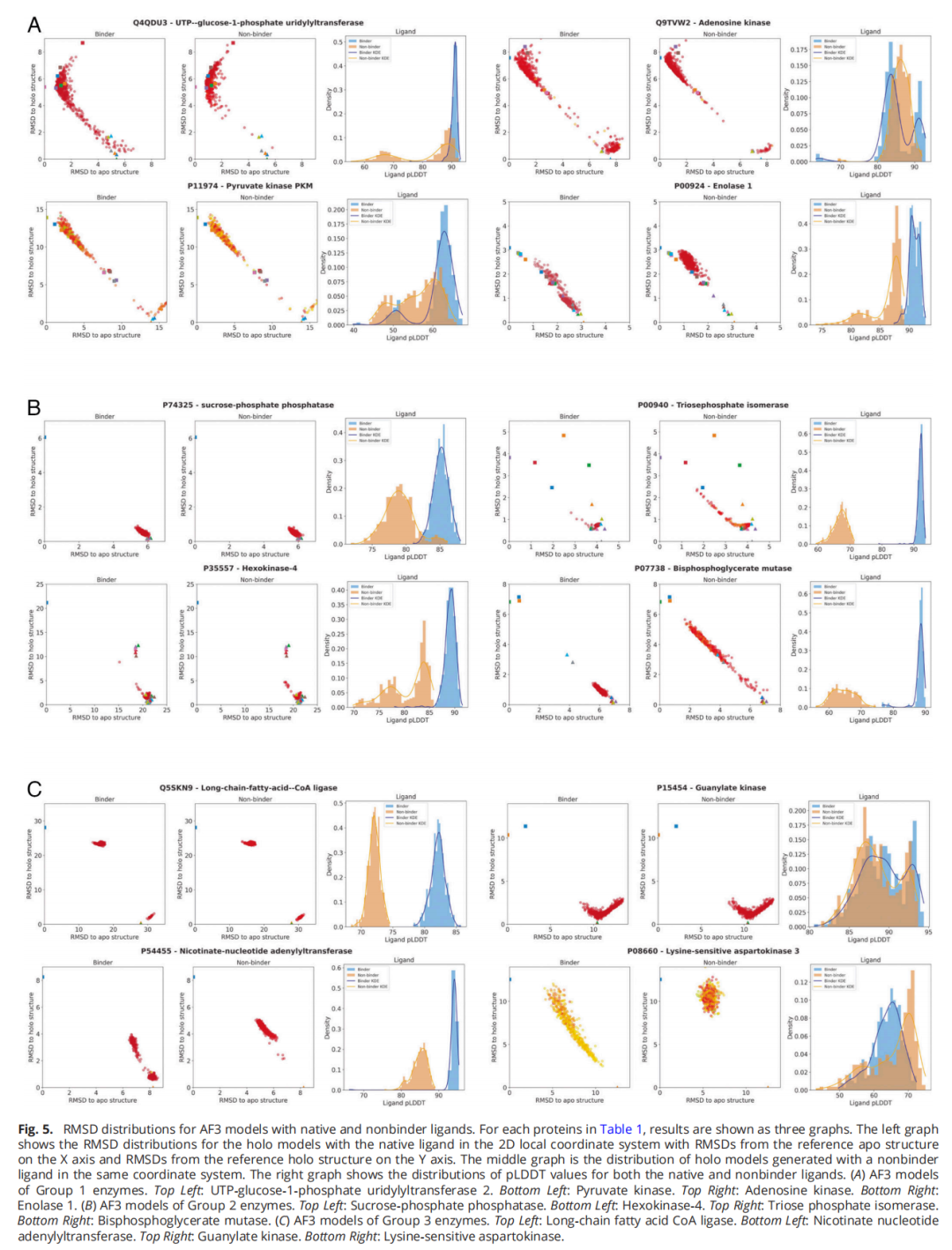
4.3 发现三:非结合配体也能诱导类似的构象变化
研究者针对 Table 1 中的 12 种酶,分别选取了文献中已知不会结合的小分子,重复了 AF3 共折叠计算。结果令人惊讶:
- • Group 2(holo 占优):非结合配体同样诱导了接近 holo 的构象,虽然部分蛋白的闭合程度略有下降。非结合配体的 pLDDT 值显著低于天然配体,但构象分布本身非常相似。
- • Group 1(apo 占优):非结合配体产生的 holo 模型比天然配体更少,对部分酶(如 enolase 1)差异较大。但 pLDDT 分布的变化很小,意味着非结合配体几乎以与天然配体相同的置信度被放置。
- • 特别案例——长链脂肪酸 CoA 连接酶:非结合物油酸的分子量远大于天然触发配体 ANP,但 AF3 仍然生成了非常相似的 holo 模型。这一案例有力地说明,配体的放置主要由结构记忆而非相互作用物理驱动。
关于 pLDDT 区分能力的结论:虽然非结合配体的 pLDDT 值总体略低于天然配体,但这种差异通常不足以可靠地区分结合物与非结合物,特别是对于 Group 1 的蛋白。这对将 AF3 的 pLDDT 作为虚拟筛选打分标准的做法提出了警告。
4.4 发现四:AF2 表现出相同甚至更强的记忆化
AF2 无法显式处理配体,但研究者仍然用相同的协议生成了 500 个 AF2 模型进行比较:
- • Group 1:AF2 模型聚集于 apo 侧,与 AF3 无配体结果相似,但更多地向 holo 方向延伸。
- • Group 2:AF2 模型几乎全部聚集在 holo 参考结构附近,表现出比 AF3 更强的偏向。
- • Group 3:AF2 与 AF3 结果高度一致。
结论:AF2 和 AF3 同样受到记忆化的影响,这不是某个特定模型版本的问题,而是基于 PDB 训练的深度学习结构预测方法的系统性特征。
五、代表性案例详解
论文在 Table 1 中列出了 12 种酶的详细分析,以下摘取几个最具说明性的案例:
5.1 己糖激酶-4(Hexokinase-4, Group 2, apo:holo = 1:28)
己糖激酶-4 催化 ATP 的磷酸基团转移至葡萄糖。在缺少葡萄糖时,该酶主要存在于超开放构象。然而,由于 PDB 中有 28 个 holo 结构但仅 1 个 apo 结构,AF3 在完全没有葡萄糖输入的情况下,全部 500 个模型都更接近闭合态。加入葡萄糖后,模型进一步移向更闭合的 holo 亚群。配体 RMSD 平均低于 1 Å——但这完全归功于结合位点已"默认"闭合,而非 AF3 真正理解了葡萄糖的结合。
5.2 丙酮酸激酶(Pyruvate Kinase, Group 1, apo:holo = 8:4)
丙酮酸激酶催化丙酮酸被 ATP 磷酸化。500 个无配体模型聚集在两组 apo X 射线结构之间。加入 ATP 后,仅 63 个模型移向 holo 侧,大部分维持原位。少量 holo 模型甚至出现部分解折叠,远离 apo 和 holo 构象,置信度低。这表明对 apo 数据占优的蛋白,配体的影响有限。
5.3 磷酸丙糖异构酶(TIM, Group 2, apo:holo = 5:13)
TIM 在 apo 态有 5 种不同构象(活性位点附近两个环的构象多样性),但与 13 个闭合 holo 结构明确可区分。AF3 在无配体的情况下,全部 500 个模型聚集在闭合 holo 结构附近。加入触发配体 PGA 后,模型更加靠近 holo,配体 RMSD < 1 Å。这是记忆化效应的经典演示。
5.4 赖氨酸敏感天冬氨酸激酶3(Lysine-sensitive aspartokinase 3, Group 3, apo:holo = 1:1)
这是一个有趣的"错误标注"案例。DynDom 数据库将 ADP 错误标记为触发配体,但实际的构象变化由别构抑制剂赖氨酸(LYS)引起。基于错误信息,AF3 在无配体和有 ADP 时都产生了介于开放和闭合态之间的宽泛分布,配体置信度低。这说明在训练数据稀少且配体不正确时,AF3 缺乏将构象推向明确状态的能力。
六、讨论与启示
6.1 "程序在建模什么?"
论文引用了 Agarwal & McShan(2024)的警示:当机器学习方法远离生物物理学时,我们可能不太清楚程序实际在建模什么。AF2 的作者早已指出,由于网络同时在 apo 和 holo 结构上训练,模型可能与配体存在时的结构一致,即使 AF2 的输入中没有配体。更一般地说,模型倾向于产生训练数据中占主导的构象,但从 pLDDT 或 PAE 图中很难判断模型究竟更像 apo 还是 holo 形式。
AF3 的共折叠能力理论上可以消除这种不确定性——你可以显式地分别建模 apo 和 holo。然而本文的结果表明,这种"显式建模"的效力远弱于训练数据分布本身的驱动力。
6.2 对药物设计和虚拟筛选的影响
本文的发现对将 AF3 应用于药物设计具有直接影响:
- 1. 对接精度的"虚假繁荣":对于 PDB 中 holo 结构丰富的蛋白(Group 2),AF3 确实能以 <2 Å 精度放置配体,但这更多反映了训练数据的记忆而非对相互作用的理解。对于 holo 结构较少的蛋白(Group 1),对接精度大幅下降。
- 2. 虚拟筛选的假阳性风险:非结合配体也能诱导类似构象变化,且 pLDDT 差异不足以可靠区分,意味着直接用 AF3 进行虚拟筛选可能产生大量假阳性。
- 3. 构象选择性的不可靠性:无法确信 AF3 预测的"闭合态"真正反映了配体结合效应,还是仅仅是训练数据偏差的产物。
6.3 训练数据量少时配体影响更大
一个积极的发现是:Group 3 蛋白(训练数据稀少)的配体效应最为显著(+17.5%),说明当记忆偏差较弱时,AF3 确实能在一定程度上利用配体信息。这暗示,对于 PDB 中结构稀少的新靶点,AF3 的共折叠预测可能比对"常见"蛋白更有参考价值——但同时也伴随更高的不确定性(如 aspartokinase 3 案例所示)。
6.4 对 AI for Science 的广泛启示
本文的意义超越了蛋白质结构预测本身。它提示我们:
- • 数据驱动方法的偏差可能被隐藏:当训练集中某类样本占优时,模型输出可能反映数据分布而非物理规律,且这种偏差在单一案例中难以察觉,需要系统性评估才能暴露。
- • "正确"的结果未必基于"正确"的原因:AF3 对 Group 2 蛋白的配体放置精度很高,但原因并非它理解了结合,而是训练集的偏差恰好有利于此。
- • 显式输入不等于显式建模:仅仅将配体作为输入提供给 AF3,并不意味着模型会物理地"考虑"配体的作用。网络的隐式偏好可能压过显式输入的影响。
七、总结
问题 | 回答 |
|---|---|
AF3 能否预测配体诱导的构象变化? | 能力有限,结果严重依赖训练数据中 apo/holo 的比例 |
记忆化效应有多强? | 训练集偏差的影响(40.3%)是配体实际影响(9–12%)的 3.5–4.5 倍 |
配体放置精度如何? | holo 结构丰富的蛋白较好(80% < 2Å),apo 占优的蛋白较差(42% < 2Å) |
非结合配体是否也诱导构象变化? | 是,且构象分布与天然配体相似 |
pLDDT 能否区分结合物与非结合物? | 通常不足以可靠区分 |
AF2 是否有相同问题? | 是,记忆化程度甚至更强 |
训练数据少时结果如何? | 配体影响更显著,但不确定性也更高 |
一句话总结:AF3 在预测酶的配体诱导构象变化时,更多地是在"回忆"训练数据中占主导的结构类型,而非真正在"预测"配体的作用——这种记忆偏见在 AF2 中同样存在,对药物设计和构象预测的可靠性提出了重要警告。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-29,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号