Nat. Mach. Intell. | 基于二维几何模板扩散的单序列蛋白结构预测增强方法

Nat. Mach. Intell. | 基于二维几何模板扩散的单序列蛋白结构预测增强方法

DrugAI
发布于 2026-04-13 15:43:34
发布于 2026-04-13 15:43:34
DRUGONE
单序列蛋白结构预测因无需依赖同源信息而具有重要应用价值,但其精度长期受限。研究人员提出TDFold,一种基于二维几何模板扩散的新方法,通过生成高质量的残基间几何关系(包括距离与取向),实现高效且准确的三维结构预测。该方法包含两个阶段:二维几何模板生成与序列-几何协同学习。实验结果表明,TDFold在多个基准数据集上优于现有蛋白语言模型方法,并在缺乏同源信息的场景中表现出显著优势,同时具有更低的计算资源需求和更高的推理效率,为单序列结构预测提供了一种新的解决方案。

近年来,人工智能在蛋白结构预测领域取得了突破性进展,以AlphaFold和RoseTTAFold为代表的方法在CASP竞赛中取得了接近实验精度的表现。然而,这类方法高度依赖多序列比对和结构模板信息,当面对缺乏同源序列的蛋白(如孤儿蛋白或快速进化的病毒蛋白)时,预测性能显著下降。
与此同时,基于蛋白语言模型的方法尝试仅利用序列信息进行预测,虽然提高了效率,但仍存在计算开销大和精度不足的问题。尤其是其核心模块通常具有较高时间复杂度,在长序列情况下难以扩展。
研究人员指出,当前方法的关键瓶颈在于缺乏对残基间几何关系的高质量建模。如果能够直接生成可靠的几何约束信息,就有可能在无需同源信息的情况下显著提升结构预测性能。
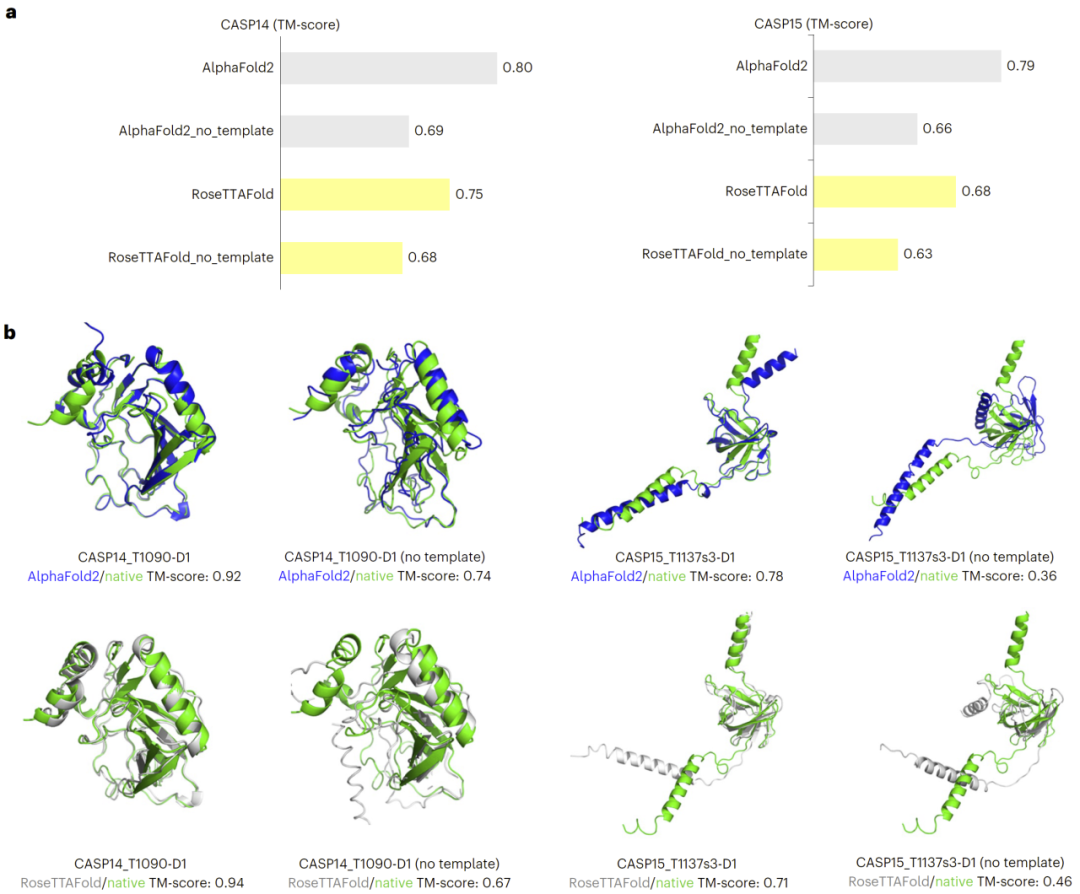
图1: 三维结构模板信息对AlphaFold2和RoseTTAFold在CASP14(N = 32)与CASP15(N = 45)数据集上预测性能的影响。
方法
研究人员提出TDFold框架,将视觉生成模型引入蛋白结构预测。首先,通过二维几何模板扩散模块,将蛋白序列作为“文本提示”,利用扩散模型生成残基间距离和取向矩阵;随后,在序列-几何协同学习模块中,结合残基级与原子级图神经网络,对结构进行精细建模并输出三维坐标。该方法通过LoRA微调策略将预训练扩散模型迁移至蛋白几何生成任务,实现序列与几何信息的高效融合。
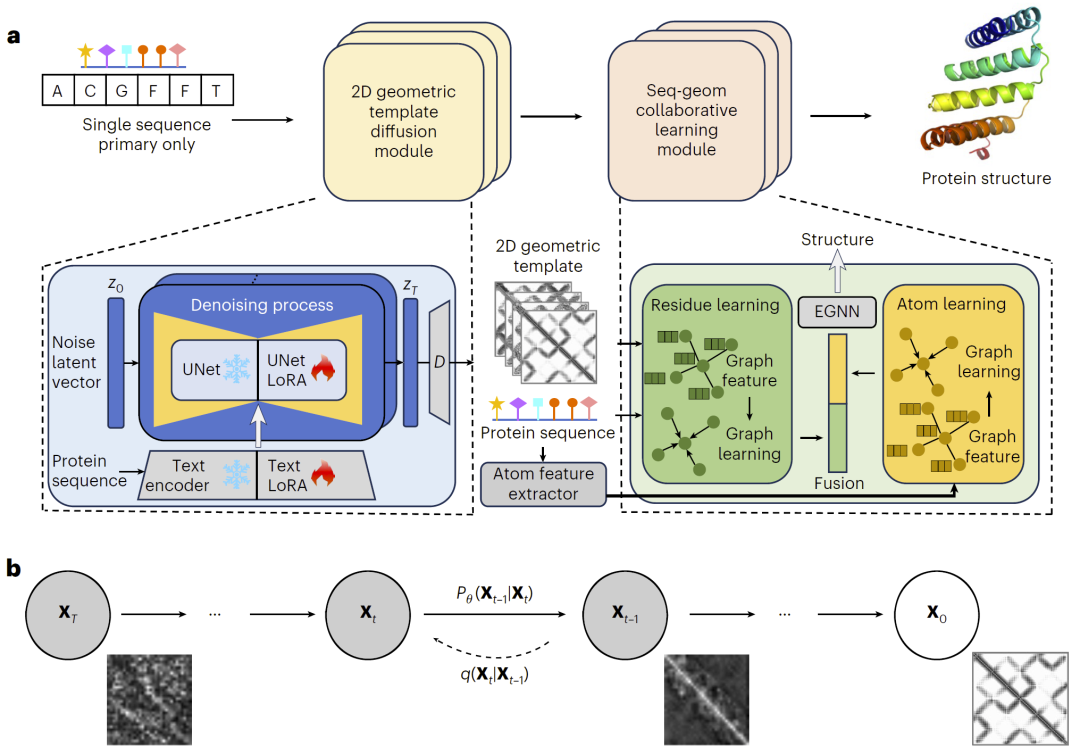
图2: TDFold的整体架构及二维几何模板扩散过程。
结果
模型整体框架与生成机制验证
研究人员首先展示了TDFold的整体架构及其扩散生成过程。模型通过逐步去噪生成二维几何模板,再结合图神经网络完成三维结构预测。这一设计实现了从序列到几何再到结构的逐级建模路径,有效弥补了传统方法中几何信息不足的问题。
在孤儿蛋白上的结构预测表现
在缺乏同源信息的Orphan和Orphan25数据集上,TDFold显著优于AlphaFold、RoseTTAFold以及多种蛋白语言模型方法。研究人员发现,即使在完全无MSA信息的情况下,TDFold仍能取得最高的TM-score和GDT_TS表现,表明其能够有效从单序列中提取结构信息。
同时,二维几何模板的可视化结果显示,TDFold生成的残基间距离矩阵更接近真实结构,相比数据库检索得到的模板具有更高准确性,从而直接提升最终结构预测性能。
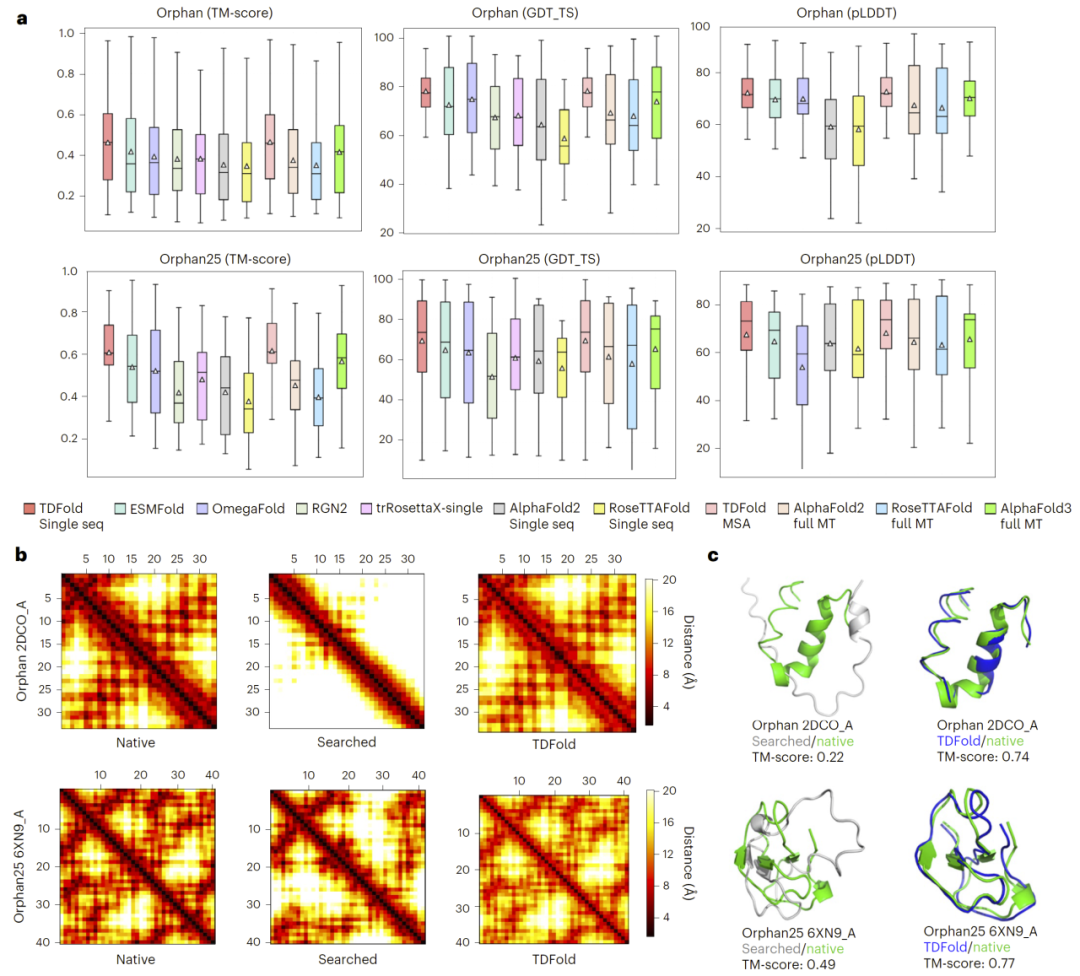
图3: 在Orphan(N = 77)与Orphan25(N = 25)数据集上的性能比较及可视化示例。
在CASP基准数据集上的性能
在CASP14、CASP15和CASP16等标准基准测试中,TDFold表现出与现有主流方法相当甚至更优的性能。与仅依赖序列的模型相比,TDFold consistently取得更高的TM-score;与依赖同源信息的方法相比,在单序列模式下优势更加明显。
研究还表明,传统方法在移除模板信息后性能显著下降,而TDFold无需依赖此类信息即可维持稳定表现,说明其几何建模能力具有较强泛化性。
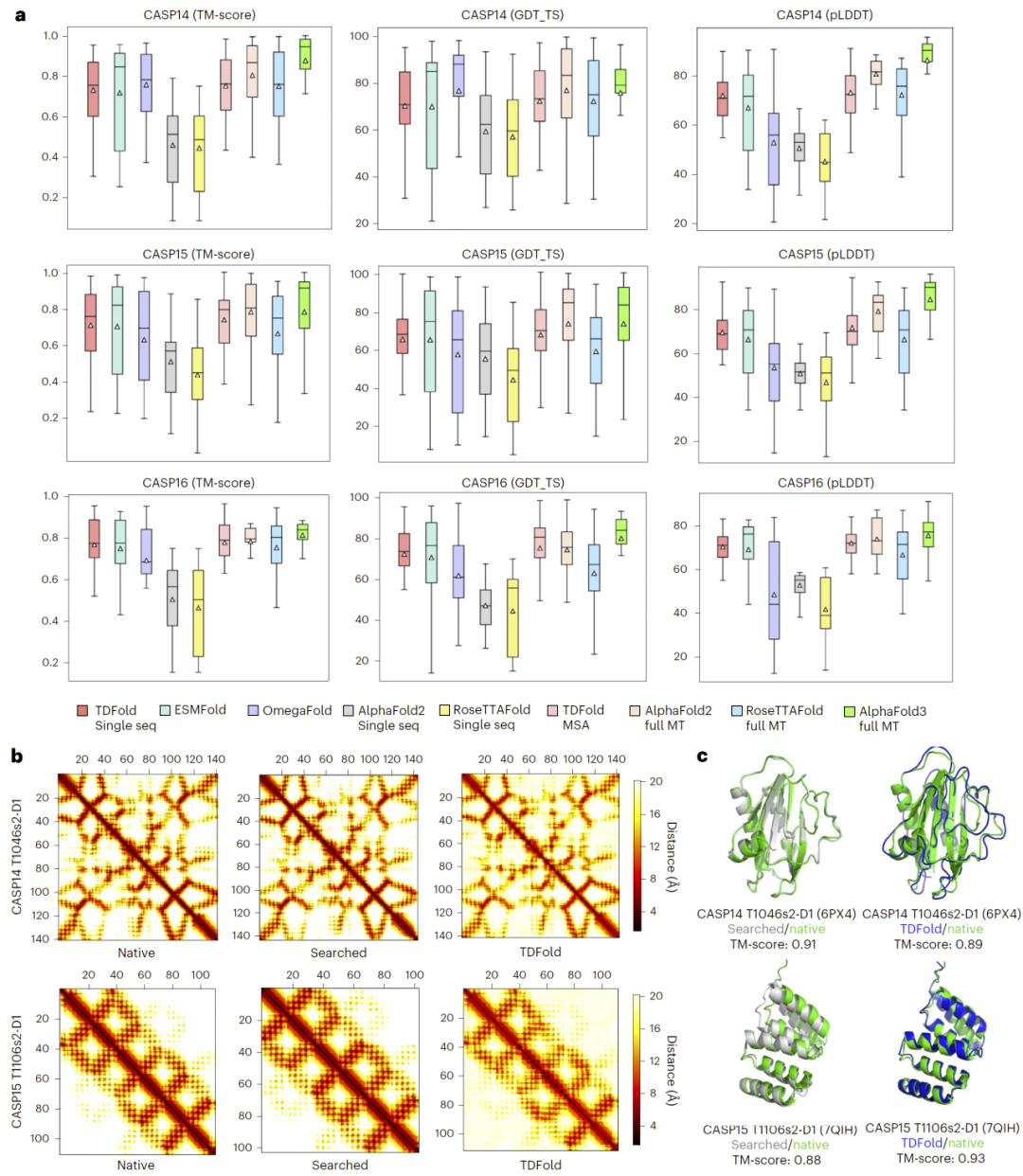
图4: 在CASP14(N = 32)、CASP15(N = 45)与CASP16(N = 15)数据集上的性能比较及可视化示例。
在病毒蛋白等困难目标上的表现
针对同源信息极少的病毒蛋白,TDFold表现出显著优势。研究人员在多个病毒相关结构预测任务中发现,该方法优于AlphaFold、ESMFold等模型,能够更准确地恢复结构特征。这对于理解病毒免疫逃逸机制和药物设计具有重要意义。
推理效率与资源消耗分析
研究人员对比了多种方法的推理时间与GPU内存消耗。结果显示,TDFold在预测一个500残基蛋白时仅需约10秒,显著快于ESMFold(约100秒)以及AlphaFold系列方法(数百至上千秒)。同时,其内存占用明显更低,仅约7GB。
这一优势源于扩散模型与轻量图网络的设计,使其计算复杂度与序列长度解耦,从而在长序列预测中具有更好的扩展性。
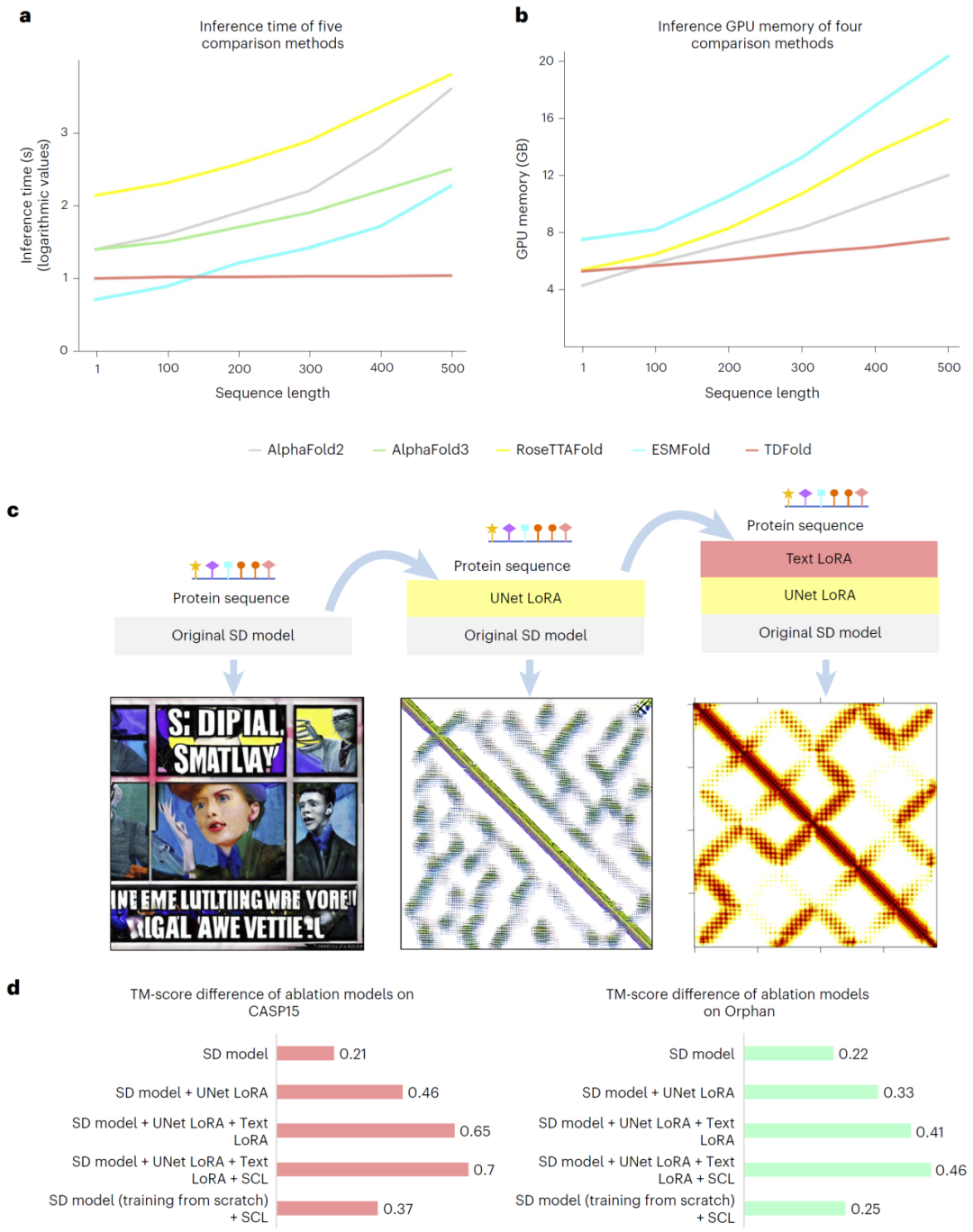
图5: 计算时间与内存开销对比及模型消融分析。
讨论
研究人员提出的TDFold方法为单序列蛋白结构预测提供了一种新的技术路径,通过引入二维几何模板扩散,有效弥补了传统方法在几何建模方面的不足。
该方法的核心创新在于将视觉生成模型与蛋白结构预测相结合,实现了从序列到几何再到结构的分层建模。同时,其无需依赖同源信息的特性,使其在孤儿蛋白和病毒蛋白等困难任务中具有显著优势。
未来,该框架有望进一步扩展至更复杂的生物体系,并与更先进的生成模型结合,从而推动蛋白结构预测向更高精度与更高效率发展。总体而言,该研究展示了一种融合生成模型与结构生物学的新范式,为资源受限环境下的蛋白研究提供了重要工具。
整理 | DrugOne团队
参考资料
Wang, X., Zhang, T., Cui, Z. et al. Two-dimensional geometric template diffusion for boosting single-sequence protein structure prediction. Nat Mach Intell (2026).
https://doi.org/10.1038/s42256-026-01210-2

内容为【DrugOne】公众号原创|转载请注明来源
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-03,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号