文献解读-Cross-kingdom metagenomics:同一批数据解析细菌、真核生物与病毒的群落特征

文献解读-Cross-kingdom metagenomics:同一批数据解析细菌、真核生物与病毒的群落特征

用户1075469
发布于 2026-05-07 16:07:17
发布于 2026-05-07 16:07:17
Hello,Hello,小伙伴们大家好!今天和大家分享一篇很有代表性的宏基因组研究文章。这项研究由山东大学相关团队开展,研究对象是我国潮间带泥滩生态系统,文章题目为:《Unveiling the biodiversity of large DNA viruses in intertidal mudflats via metagenomics》。
该研究以大型 DNA 病毒为切入点,系统解析了潮间带泥滩中 giant viruses 和 large phages 的多样性、系统发育关系、生态分布格局和功能潜力。研究共整合了 199 个沉积物宏基因组,数据量约 5.3 TB,并通过病毒序列识别、基因组延伸、viral binning、质量评估和去冗余等步骤,从中恢复出 237 个大型 DNA 病毒 vMAGs,展示了潮间带这一复杂动态环境中尚未被充分认识的病毒多样性。不过,这篇文章的亮点并不只是“发现了多少病毒、如何分类”。更重要的是,研究者将这些大型 DNA 病毒放回生态系统背景中,进一步分析它们与潜在宿主、环境因子以及物质循环之间的关系。
从分析框架来看,研究者并没有把病毒作为孤立对象来分析,而是进一步结合了真核生物群落解析、细菌宿主预测、病毒-宿主关联网络、生态组装过程和代谢功能注释等多个层面的结果,将大型 DNA 病毒放回到更完整的生态系统背景中进行解释。其中,真核生物分析是这篇文章跨界框架中非常关键的一环。研究者从同一批宏基因组数据中识别 eukaryotic contigs,并进一步解析潮间带泥滩中的真核生物组成,包括藻类、动物、原生生物和真菌等类群。随后,研究结合真核生物丰度信息与 giant viruses 的分布特征,通过相关性分析和共现网络,推测巨型病毒与潜在真核宿主之间的生态关联。
因此,这篇文章非常适合作为 Cross-kingdom metagenomics 的一个案例。它展示了同一批宏基因组数据如何被拆解成不同的生态信息层:一方面可以通过 bacterial MAGs 和宿主预测方法解析 large phages 与细菌之间的潜在联系;另一方面可以通过 eukaryotic contig 识别和真核群落分析,探索 giant viruses 与真核生物之间的关联;同时还可以进一步讨论病毒在碳、氮、硫等元素循环中的潜在作用。
对于从事宏基因组分析的研究者而言,这篇文章提供的不只是一个大型 DNA 病毒分析流程,更是一种值得借鉴的研究思路:不要只把宏基因组数据看作细菌数据,而应尝试从细菌、真核生物和病毒三个层面共同理解一个生态系统。
摘要
大型 DNA 病毒(large DNA viruses, LDVs) 是地球病毒圈中一类独特的病毒类群,以 超大的基因组 和 广泛的代谢潜力 为主要特征。然而,目前对该类病毒的认识仍十分有限,尤其是在 复杂且高度动态的生态环境 中。本研究采用多种采样与测序策略,基于总量达 5.3 TB 的宏基因组数据,从 潮间带泥滩 中系统恢复了 237 个宏基因组组装的 LDV 基因组。系统发育分析在 Imitervirales 中识别出一个具有 独特演化地位的亚群,该亚群与 多种真核生物谱系 存在广泛关联。部分 LDV 种群 能够在局部环境中持续存在,并表现出 显著的基因组变异,这些变异可能与 潮间带环境的动态变化 有关。生态学分析从 群落和遗传两个层面 揭示了 LDVs 的分布格局:与大型噬菌体相比,巨型病毒的群落周转更为剧烈,但其 核苷酸多样性的变化幅度相对较弱。此外,LDVs 与其潜在宿主呈现出 相似的宏生态格局,而这些宿主也在很大程度上塑造了 LDV 群落的组装过程。潮间带 LDVs 编码了 多样的功能基因,但其中多数仍缺乏明确的功能注释;通过引入 蛋白语言模型,未知噬菌体基因的功能注释率提高了 27.32%。尽管 巨型病毒和大型噬菌体 在功能基因组成上较为相似,但二者在 特定代谢通路 上表现出不同偏好,尤其是与 碳循环和氮循环 相关的通路。该研究加深了对 潮间带这一研究不足生态系统 中 LDVs 生物多样性、生态格局及功能潜力 的认识。
材料与方法
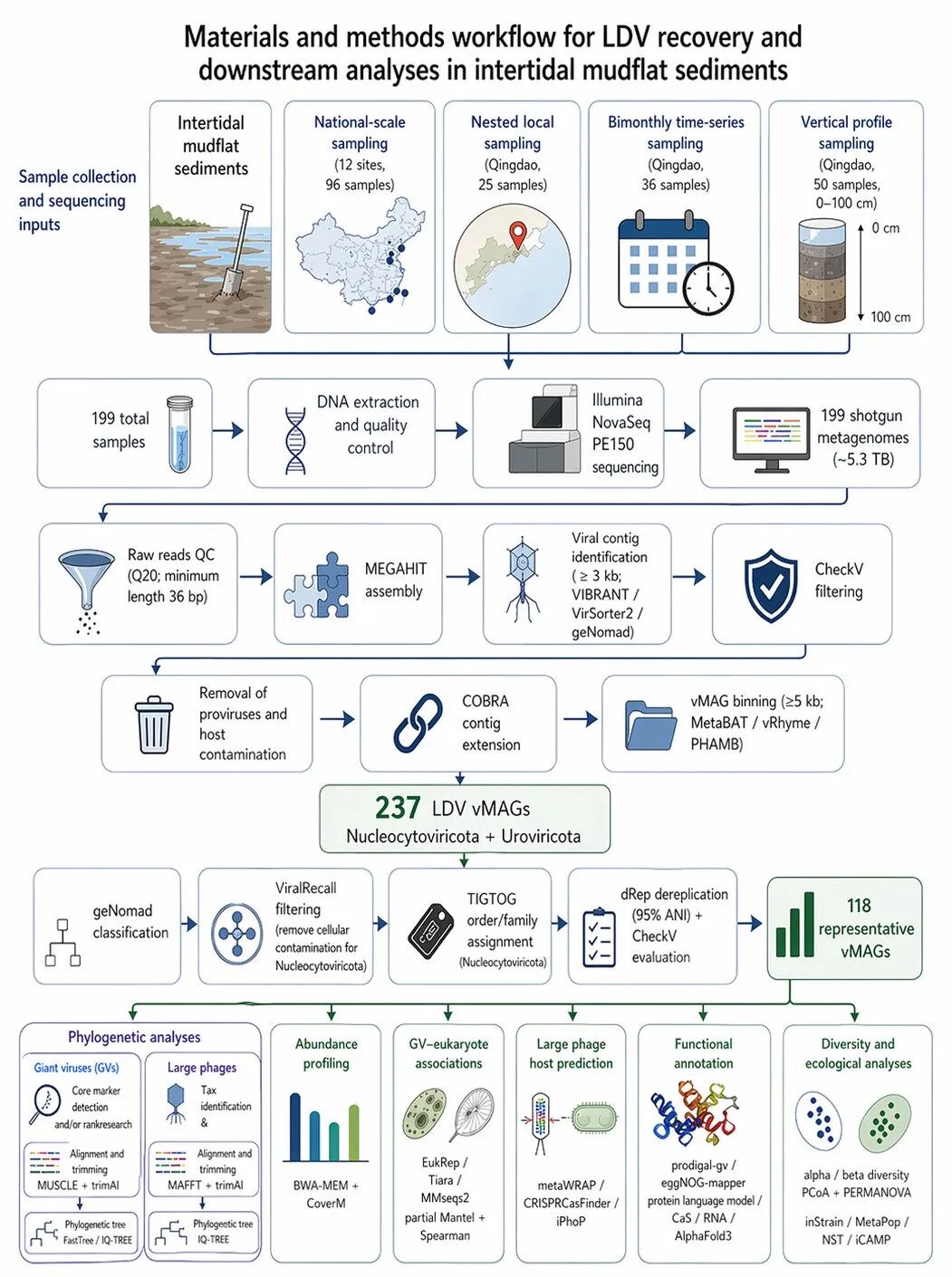
图|潮间带泥滩沉积物中大型 DNA 病毒(LDVs)的宏基因组重建与下游分析流程。
本研究通过全国尺度采样、局域嵌套采样、双月时间序列采样和垂直剖面采样,共获得 199 个潮间带沉积物样品。所有样品经 DNA 提取、质量控制和 Illumina NovaSeq PE150 测序后,获得约 5.3 TB 鸟枪法宏基因组数据。
随后,对 raw reads 进行质控并使用 MEGAHIT 组装;组装得到的 contigs 进一步结合 VIBRANT、VirSorter2、geNomad 和 CheckV 进行病毒序列识别与质量筛选。经前病毒序列和宿主污染去除、COBRA contig 延伸,以及 MetaBAT、vRhyme 和 PHAMB binning 后,共重建得到 237 个 LDV vMAGs。
进一步通过 geNomad 分类、ViralRecall 污染过滤、TIGTOG 分类注释、dRep 去冗余和 CheckV 质量评估,筛选得到 118 个代表性 vMAGs。基于这些代表性基因组,研究进一步开展系统发育分析、丰度分析、GV–真核生物相关性分析、大型噬菌体宿主预测、功能注释、多样性分析和生态学分析,以解析潮间带 LDVs 的生物多样性、系统发育关系、宿主关联、功能潜力及群落组装机制。
主要结果
结果1|潮间带环境中大型DNA病毒基因组的组装与特征解析
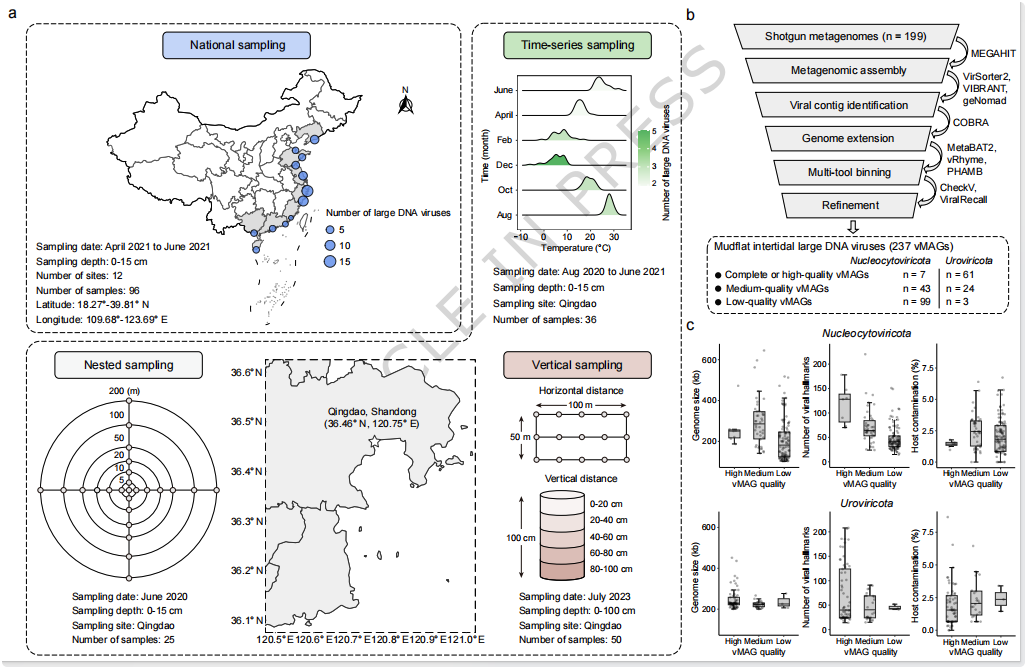
本研究基于我国沿海潮间带泥滩沉积物的多尺度采样与宏基因组测序数据,共获得约 5.3 TB 数据(Fig. 1a),为大型DNA病毒的系统挖掘提供了数据基础。研究构建了一套较完整的病毒基因组恢复流程(Fig. 1b):先利用 VirSorter2、VIBRANT 和 geNomad 识别病毒序列,再结合 COBRA 进行序列延伸,并通过 MetaBAT2、vRhyme 和 PHAMB 等方法进行病毒 binning。最终共恢复得到 237 个大型DNA病毒基因组(vMAGs),包括 149 个巨型病毒(Nucleocytoviricota) 和 88 个大型噬菌体(Uroviricota)。其中,15 个为完整基因组,53 个为近完整基因组(>90% 完整度),显示出较高的基因组恢复质量。进一步的基因组特征分析表明(Fig. 1c),巨型病毒基因组大小为 103–645 kb,大型噬菌体为 200–452 kb;同时,基因组完整性较高的 vMAGs 通常携带更多病毒标志基因、宿主污染更低。基于 95% ANI 聚类,这些 LDVs 可归并为 118 个代表性物种,说明潮间带环境中蕴藏着丰富且尚未被充分认识的大型DNA病毒多样性。
结果2|潮间带环境中大型DNA病毒的系统发育多样性解析
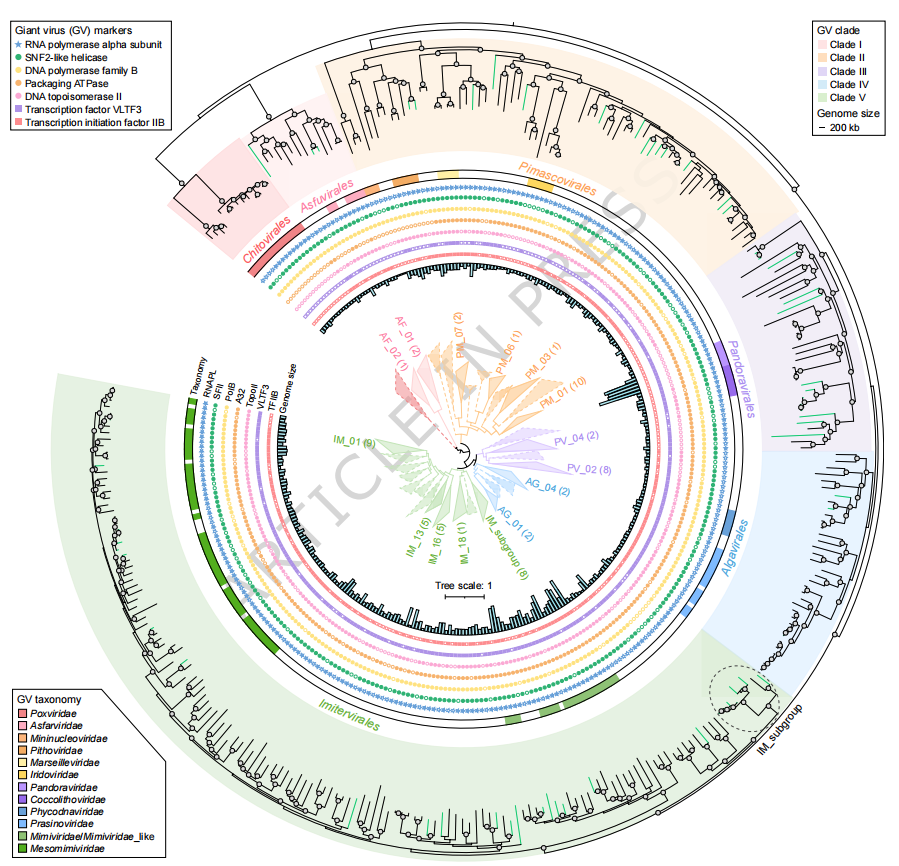
在获得高质量病毒基因组的基础上,研究进一步基于保守标志基因构建系统发育树,对潮间带大型DNA病毒的进化多样性进行解析(Fig. 2)。结果显示,潮间带巨型病毒主要分布于 Imitervirales、Pimascovirales、Pandoravirales、Algavirales 和 Asfuvirales 五个病毒目,其中 Imitervirales 为优势类群;同时,与典型海洋环境相比,潮间带中 Algavirales 相对较少,而 Pimascovirales 呈现一定富集,反映出潮间带这一陆海过渡生态系统的独特病毒组成。进一步分析发现,这些巨型病毒可归属于多个已知家族(如 Mimiviridae、Mesomimiviridae、Asfarviridae 等),同时仍有相当一部分聚类于尚未命名的分支,显示出较高的未知多样性。值得注意的是,研究在 Imitervirales 内鉴定到一个由 8 个潮间带巨型病毒基因组组成的独立分支,该分支在系统发育上与已知亚群相邻但明显分化,并在平均氨基酸相似性(AAI)上表现出较高的类群内一致性,提示其可能代表一个尚未被定义的 family-level 新类群。总体来看,潮间带环境中的巨型病毒在系统发育上覆盖多个已知类群,并呈现出明显的谱系扩展特征,进一步丰富了当前对大型DNA病毒进化多样性的认识。
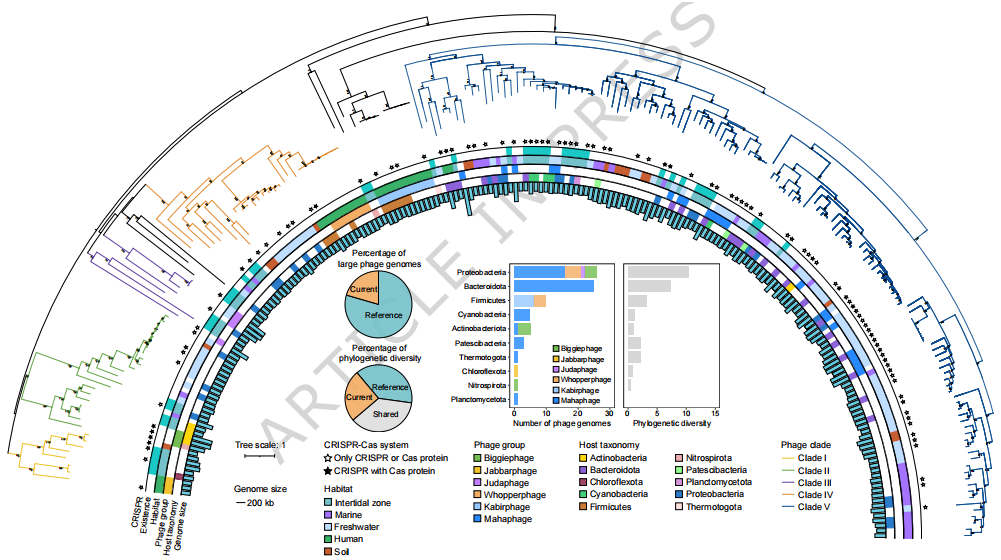
针对大型噬菌体,研究进一步基于 TerL 蛋白构建系统发育树,解析其进化分布特征(Fig. 3)。结果显示,潮间带大型噬菌体并非集中于单一类群,而是分布在多个已知大型噬菌体分支中,其中多数归属于全球多种生态系统中广泛存在的 Mahaphage 类群。同时,部分潮间带大型噬菌体与 Biggiephage、Judaphage、Jabbarphage 和 Whopperphage 等参考类群聚类在一起,说明潮间带环境进一步扩展了这些大型噬菌体类群的已知系统发育和基因组多样性。总体来看,Fig. 3 表明潮间带大型噬菌体具有较广泛的进化来源,并与多种已知大型噬菌体谱系存在密切关联。
结果3|潮间带环境中大型DNA病毒的宏生态分布格局与遗传变异特征
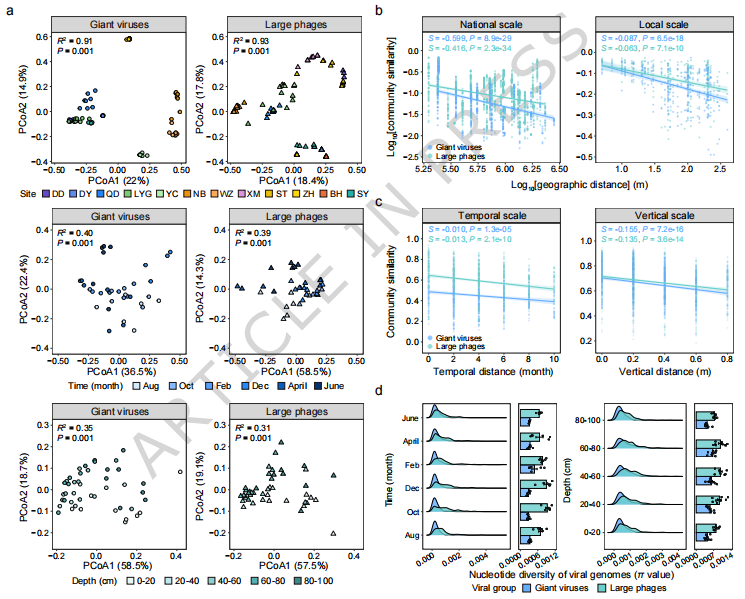
在系统发育解析的基础上,研究进一步从空间、时间和垂直维度探讨了潮间带大型DNA病毒的宏生态分布格局及其遗传变异特征(Fig. 4)。主坐标分析结果表明,病毒群落在不同样点之间呈现明显分离,且 采样地点是影响群落结构的主要因素,其次为时间和深度,说明潮间带病毒群落具有显著的空间异质性(Fig. 4a)。进一步分析发现,大型DNA病毒群落普遍遵循典型的 距离衰减关系(distance–decay relationship, DDR),即地理距离越远,群落相似性越低(Fig. 4b);其中,巨型病毒的衰减斜率更陡,表明其群落更易发生空间替代。除空间尺度外,病毒群落在时间和沉积物深度方向上同样表现出一定程度的更替(Fig. 4c),反映出潮间带环境动态变化对病毒群落的持续影响。
在遗传变异层面,研究通过核苷酸多样性(nucleotide diversity)评估不同病毒类群的微观进化特征。结果显示,大型噬菌体在不同尺度上均表现出 更高的核苷酸多样性及更大的变异幅度,而巨型病毒的遗传多样性相对较低且更稳定(Fig. 4d)。这一差异可能与其宿主类型、复制方式以及进化约束有关,例如巨型病毒依赖真核宿主、复制周期较长,从而积累的突变较少。总体来看,Fig. 4 揭示了潮间带大型DNA病毒在宏观生态格局和微观遗传变异上的双重特征,体现了其对复杂环境的响应及不同类群间的进化策略差异。
结果4|潮间带环境中大型DNA病毒与潜在宿主的关联模式及生态指示
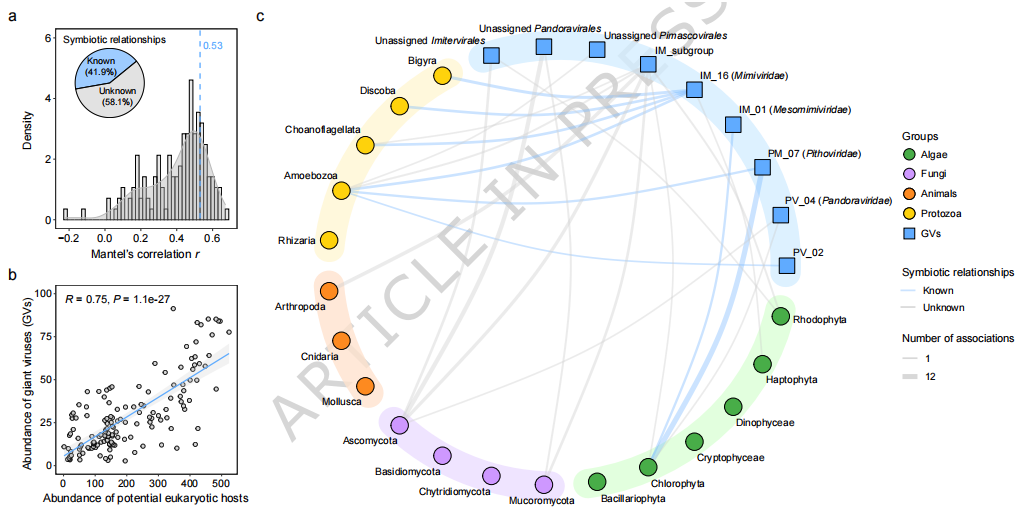
在宏生态格局分析的基础上,研究进一步探讨了潮间带大型DNA病毒与潜在宿主之间的关联特征(Fig. 5)。通过对沉积物中真核生物群落的解析,共识别出包括藻类、动物、原生生物和真菌在内的多类宿主类群,并发现其在不同空间尺度上具有明显的分布差异。基于控制地理距离的 Mantel 检验,共鉴定出多组显著的病毒—宿主关联关系,其中约 40% 已被已有研究报道,表明结果具有较高可靠性(Fig. 5a)。进一步的共现网络分析揭示,大型DNA病毒与潜在宿主之间存在广泛且复杂的关联结构,整体上呈现 显著正相关关系(Fig. 5b),说明病毒群落变化在很大程度上与宿主群落动态相耦合。从宿主类型来看,真菌和原生生物与巨型病毒之间的关联最为密集,其次为藻类,而动物类群关联相对较少(Fig. 5c);其中,一些此前未被明确报道的宿主类群(如部分真菌谱系)也显示出较强关联,提示其可能在潮间带生态系统中参与巨型病毒的传播或演化过程。此外,在 Imitervirales 中识别出的潜在新分支与节肢动物类群表现出较强关联,暗示该类病毒可能与动物相关宿主存在特定的生态联系。总体而言,Fig. 5 表明 宿主群落结构是塑造大型DNA病毒分布与群落组成的关键因素,为理解病毒—宿主互作及其生态功能提供了重要依据。
结果5|潮间带环境中大型DNA病毒群落组装的生态驱动机制
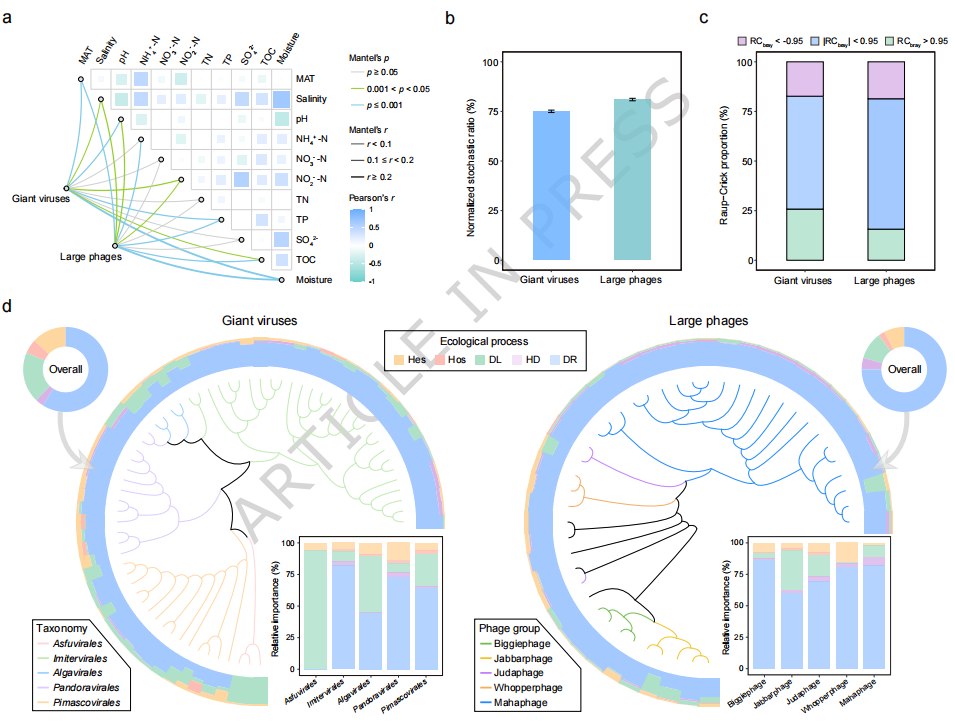
在明确病毒与宿主关联关系的基础上,研究进一步解析了潮间带大型DNA病毒群落的生态驱动因素及组装机制(Fig. 6)。通过对多种环境因子的分析发现,温度(MAT)、盐度、pH、总有机碳(TOC)以及氮、磷等营养因子均与病毒群落结构变化显著相关(Fig. 6a),表明环境梯度对病毒分布具有一定影响。然而,进一步基于空模型(null model)分析表明,随机过程(stochastic processes)在群落组装中占主导地位,对巨型病毒和大型噬菌体的贡献分别约为 75% 和 81%(Fig. 6b–c)。在具体生态过程层面,群落组装主要由 生态漂变(drift)驱动,其次为 扩散限制(dispersal limitation)和异质选择(heterogeneous selection)(Fig. 6d),说明在潮间带这一动态环境中,随机性过程对病毒群落形成具有更强控制作用。进一步分析发现,病毒群落的随机性与宿主群落显著相关,且随着宿主丰度增加,随机过程的作用强度明显降低,提示宿主不仅影响病毒分布,还通过调控种群规模间接影响群落组装机制。总体来看,Fig. 6 表明潮间带大型DNA病毒群落主要受随机过程驱动,但同时受到环境因子和宿主群落的共同调控,体现了多因素协同作用下的复杂生态组装机制。
结果6|潮间带环境中大型DNA病毒基因的代谢潜力与进化选择解析
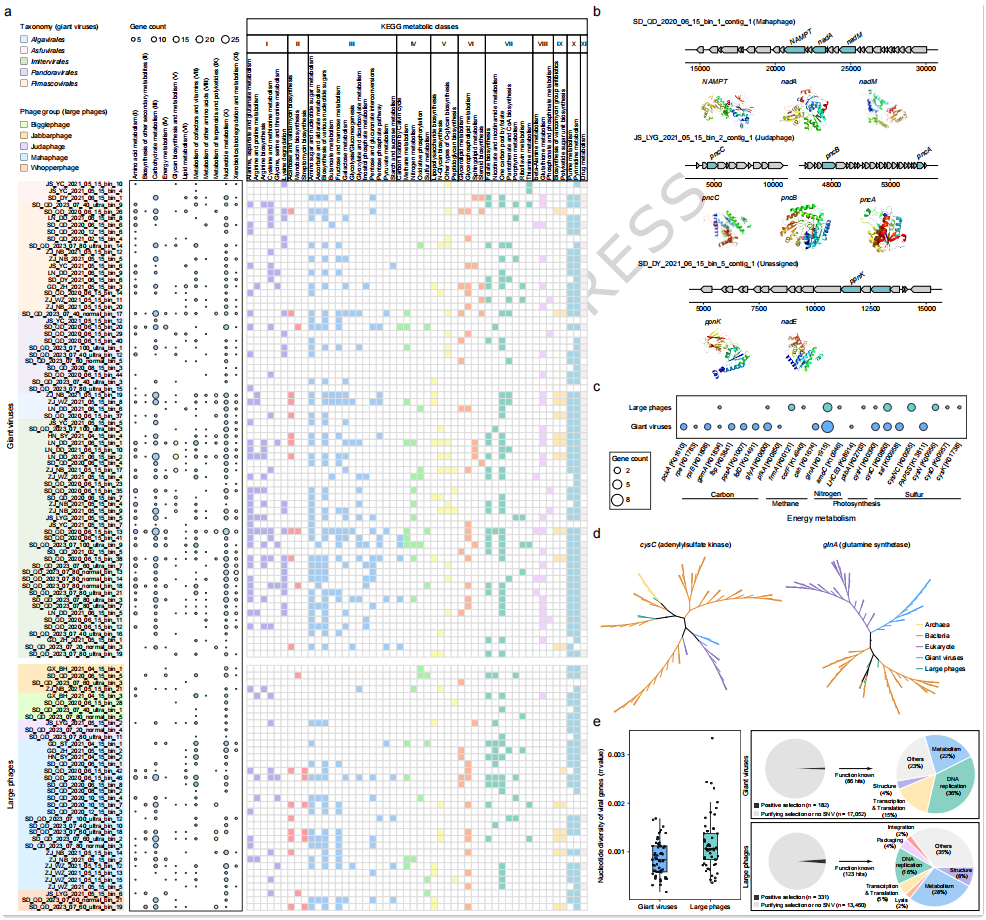
在群落组装机制分析的基础上,研究进一步探讨了潮间带大型DNA病毒的功能潜力及其进化选择特征(Fig. 7)。共预测得到大量蛋白编码基因,其中巨型病毒具有更高比例的已知功能注释,而大型噬菌体中 超过一半基因 initially 属于“功能未知”,通过蛋白语言模型进一步提升了注释率(Fig. 7a)。从代谢潜力来看,两类病毒均编码多种与 核苷酸代谢、氨基酸代谢和碳水化合物代谢 相关的基因,但 巨型病毒在代谢通路覆盖范围上更广,例如携带与 O-糖基化合成 等真核相关过程有关的基因,而大型噬菌体则更倾向于编码 NAD⁺ 合成相关通路(Fig. 7a–b)。在辅助代谢基因(AMGs)方面,研究在巨型病毒和大型噬菌体中分别鉴定出多种与 碳、氮、硫循环 相关的关键基因,如 cysC 和 glnA,表明其可能通过重编程宿主代谢参与生物地球化学过程(Fig. 7c–d)。进一步的进化分析显示,大型噬菌体整体具有 更高的核苷酸多样性,且 处于正选择(pN/pS > 1)的基因比例更高,这些基因主要涉及 DNA复制、结构蛋白及代谢相关功能(Fig. 7e),反映出病毒在与宿主相互作用过程中受到持续选择压力。总体来看,Fig. 7 揭示了潮间带大型DNA病毒不仅具备丰富的代谢潜力,还在进化层面表现出显著的适应性特征,进一步支持其在生态系统功能中的重要作用。
数据代码
- 本研究宏基因组数据已上传至 NCBI SRA,项目编号:PRJNA957716、PRJNA1029225、PRJNA1099403、PRJNA1100757、PRJNA1099773;
- 本研究组装和 binning 获得的 LDVs 代表性序列已上传至 ENA,项目编号:PRJEB108004;
- Zenodo 链接:https://zenodo.org/records/18154325;
- GitHub 链接:https://github.com/MengzhiJ/Unveiling-the-biodiversity-of-large-DNA-viruses-in-intertidal-mudflats-via-metagenomics。
学习建议
- 建议重点学习病毒 binning 与真核 contig 识别这两个核心模块
该研究最值得借鉴的,不只是大型 DNA 病毒的多样性结果,而是其较为完整的分析框架。一方面,文章系统展示了病毒序列从识别、筛选、基因组延伸、viral binning 到 vMAG 重建的完整流程;另一方面,又进一步从同一批宏基因组组装结果中识别真核生物 contigs,并将其用于 giant viruses 潜在宿主关联分析。前者关系到病毒基因组恢复的完整性与可信度,后者则决定了病毒研究能否进一步延伸到宿主背景和生态解释层面。
- 病毒分析部分的学习重点,在于理解如何从 contig-level 走向 genome-level
本文在病毒分析中并未停留在 viral contig 水平,而是通过 genome extension、multi-tool viral binning、质量评估、污染控制和去冗余等步骤,尽可能恢复更高质量的 vMAGs。因此,学习时不应只关注使用了哪些工具,更应重点理解其背后的分析逻辑:为什么大型 DNA 病毒需要 binning,为什么要联合多种工具,以及为什么 binning 后还必须经过严格的质量控制。这部分内容对于后续开展环境宏病毒研究、提升病毒基因组恢复质量具有很强的参考价值。
- 真核生物分析部分的学习重点,在于为 giant viruses 提供潜在宿主背景
作者从同一批宏基因组数据中识别真核 contigs,解析潮间带泥滩中的真核生物组成,并将这些结果与 giant viruses 的分布特征结合,用于推测潜在宿主关联。其方法学价值不只是“筛出真核序列”,而在于将真核群落信息整合进病毒生态研究之中。总体来看,这篇文章最重要的启发在于:通过 viral binning 提高大型 DNA 病毒基因组恢复质量,同时通过真核 contig 识别补充宿主背景信息,从而将病毒多样性、宿主关联和生态功能纳入同一个分析框架。
参考文献
Ji, M., Li, Y., Wang, M. et al. Unveiling the biodiversity of large DNA viruses in intertidal mudflats via metagenomics. Nat Commun. 2026 Mar 24. doi: 10.1038/s41467-026-71095-7. Epub ahead of print. PMID: 41872229.
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-24,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号