Chem. Sci. | 生成式AI探索千万级催化剂化学空间

Chem. Sci. | 生成式AI探索千万级催化剂化学空间

DrugAI
发布于 2026-05-26 20:10:01
发布于 2026-05-26 20:10:01
DRUGONE
催化材料的发现本质上是对巨大化学空间的系统探索,但传统实验与理论方法能够覆盖的范围极其有限。研究人员提出了一种基于分布式生成式 Transformer 的可扩展框架,将 Transformer 生成模型与分布式并行生成-筛选流程结合,实现了千万级催化剂结构空间的探索。通过引入降维分析与机器学习势能模型(MLP)进行性能预测,研究人员构建了一个包含超过1000万个候选结构的催化剂数据库,使可探索空间相比已有生成模型扩大了两个数量级。
在具体应用中,研究人员以甲烷转化反应中的关键中间体 CH3 为目标,通过预训练结合任务特异性微调,实现了对目标吸附结构超过90%的有效生成率。基于机器学习加速筛选,框架成功重新发现了26种已知高活性催化剂,并进一步识别出超过1200种此前未报道的潜在候选体系。进一步的 subgroup discovery 分析揭示了元素协同作用如何调控表面电子结构,从而将 CH3 吸附能调节至最优窗口。研究结果表明,该框架能够将生成式探索与高通量性能映射无缝结合,为前所未有规模的催化剂发现提供通用范式。

催化材料的开发对于化学品和能源的可持续生产至关重要,但真实催化体系对应的化学空间往往包含数千万甚至数亿种可能的组成与结构。长期以来,催化剂开发主要依赖经验驱动的“试错法”,不仅效率低,而且资源消耗巨大。高通量实验与高通量计算方法虽然在一定程度上改善了这一问题,但其探索能力仍然有限。
近年来,机器学习逐渐成为催化剂发现的重要工具。支持向量机、人工神经网络与决策树等模型能够从高维实验与计算数据中挖掘隐藏规律,从而减少对化学直觉的依赖。与此同时,生成式人工智能的发展进一步推动了材料逆向设计的出现。VAE、GAN 与扩散模型已经被用于生成沸石、半导体以及金属有机框架等结构。
Transformer 大语言模型的出现,则让研究人员能够直接基于文本或结构序列生成材料结构。在催化领域,GPT-2 架构已经被用于生成异相催化剂表面。然而,现有方法通常只能生成几十万量级的候选结构,远不足以覆盖真实催化化学空间。此外,即使生成大量结构,如何从中筛选出真正具有实际价值的催化剂仍然是巨大挑战。
为解决这些问题,研究人员提出了一种基于分布式生成式 Transformer 的大规模生成-筛选框架,旨在实现千万级催化剂结构探索,并将生成、性能预测与高通量筛选统一到一个闭环工作流中。
方法
研究人员首先使用 Open Catalyst 2020 数据集中的200万条催化剂结构,对 Transformer 生成模型进行预训练。模型采用12层 Transformer 架构,包含512维隐藏层、8个注意力头以及最大1024长度序列输入。结构信息被编码为包含晶格参数、元素种类和三维坐标的 token 序列。随后,研究人员针对 CH3 吸附结构进行微调,使模型能够定向生成与甲烷活化相关的催化剂构型。
生成完成后,研究人员利用 UMAP 降维与哈希去重方法压缩结构空间,并通过聚类识别具有代表性的局部环境。随后,使用预训练的 EquiformerV2 机器学习势能模型预测 CH3 吸附能,以此作为甲烷活化性能的第一轮描述符。最终,基于 Sabatier 原理建立最优吸附能窗口,对生成结构进行高通量筛选。
结果
构建千万级 CH3 吸附催化剂结构空间
研究人员首先分析了微调数据规模对生成效率的影响。随着用于微调的 CH3 吸附结构数量从2000增加到12000,生成成功率显著提升,并逐渐趋于稳定。最终模型对 CH3 吸附结构的生成成功率超过90%。
利用分布式生成框架,研究人员总共生成了1000万个候选结构,其中9999949个为有效原子构型,约905万包含 CH3 吸附结构。在经过原子重叠过滤后,最终保留了863万非重叠结构用于后续分析。相比已有生成框架,这一规模提升了两个数量级。
研究人员进一步利用 UMAP 对百万级结构进行二维投影,结果显示生成结构形成两个主要高密度簇,并通过连续桥接区域相互连接。这说明模型不仅能够生成大量结构,还能够覆盖连续而多样的化学空间。
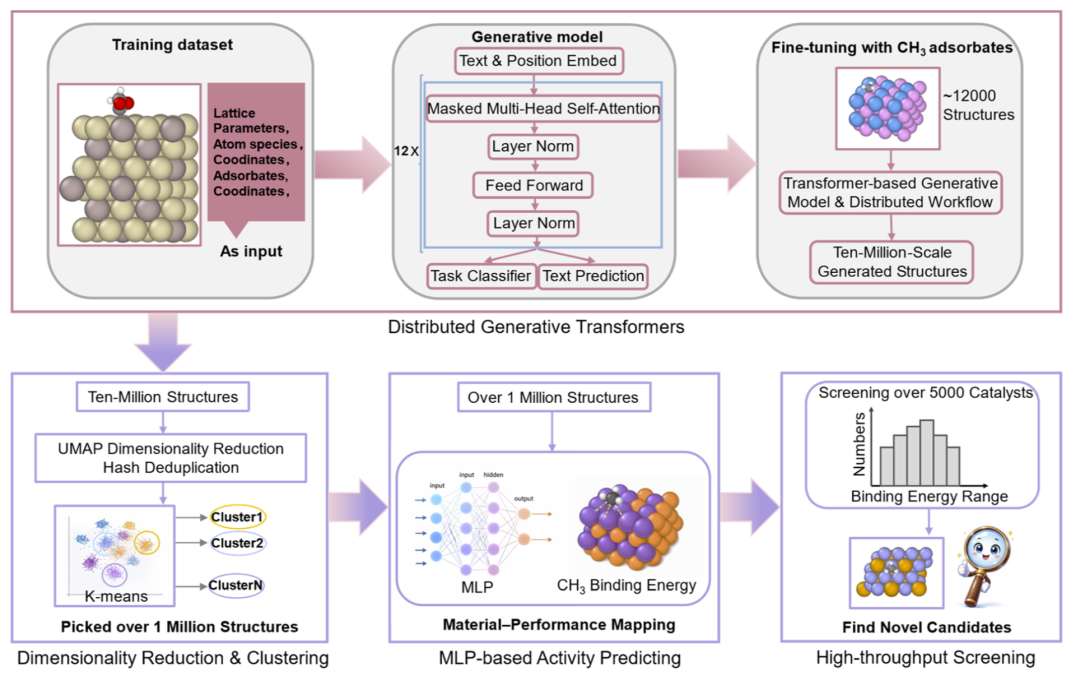
图1:分布式生成式 Transformer 工作流与千万级 CH3 吸附结构生成。
生成结构展现广泛元素多样性与连续化学空间
研究人员进一步统计了生成结构中的元素组成,并将其可视化为周期表热图。结果显示,生成体系覆盖了大量金属与非金属元素,其元素分布趋势与 OC20-S2EF 训练集高度一致。
与此同时,生成结构并非简单复制训练数据。结构嵌入分析与 SOAP 局域环境分析表明,生成结构具有更广泛的最近邻分布,并包含大量训练集中不存在的新局域环境。这意味着模型实现了“基于训练知识的化学空间探索”。
研究人员还观察到,UMAP 空间中的颜色梯度随主要元素比例和局域配位数连续变化,说明生成结构形成了连续变化的固溶体与合金体系,而非离散孤立结构。
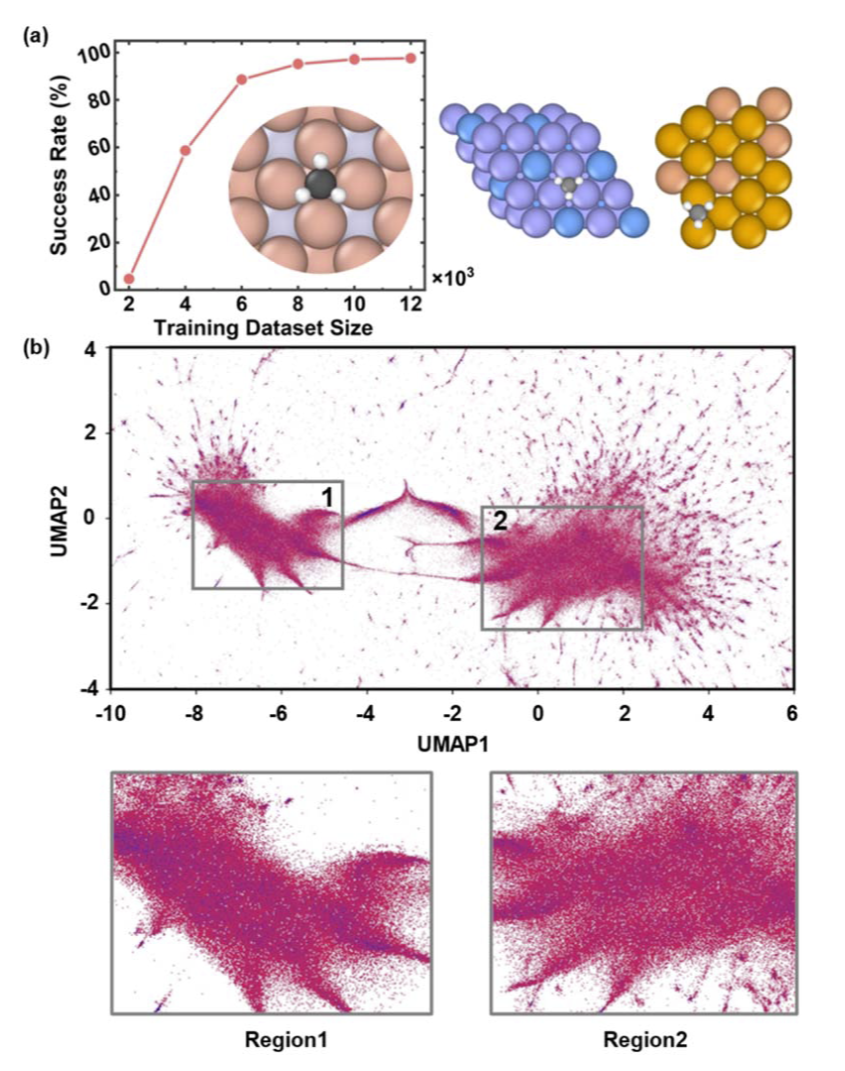
图2:生成结构的元素周期表分布与 UMAP 化学空间映射。
分层筛选实现百万级候选催化剂压缩
面对超过860万个候选结构,研究人员设计了分层筛选流程。首先通过哈希去重删除局域环境重复结构,将数据量缩减至760万。随后,基于局域环境描述符进行 k-means 聚类,得到2000个结构簇。
研究人员从每个簇随机采样1%的结构,并利用 EquiformerV2 模型预测 CH3 吸附能。随后,根据具有负吸附能结构的比例对簇进行排序,并保留前400个簇。最终约120万个结构进入完整吸附能评估流程。
结果显示,大多数 CH3 吸附能分布集中于 −1.5 至 −0.5 eV 区间,这一区域被认为是甲烷活化最优吸附能窗口。研究人员进一步发现,不同元素在不同吸附能区间中呈现明显分布差异。例如,Rh 和 Ru 更倾向出现在中等吸附能区域,而 Fe 则更集中于强吸附区域。
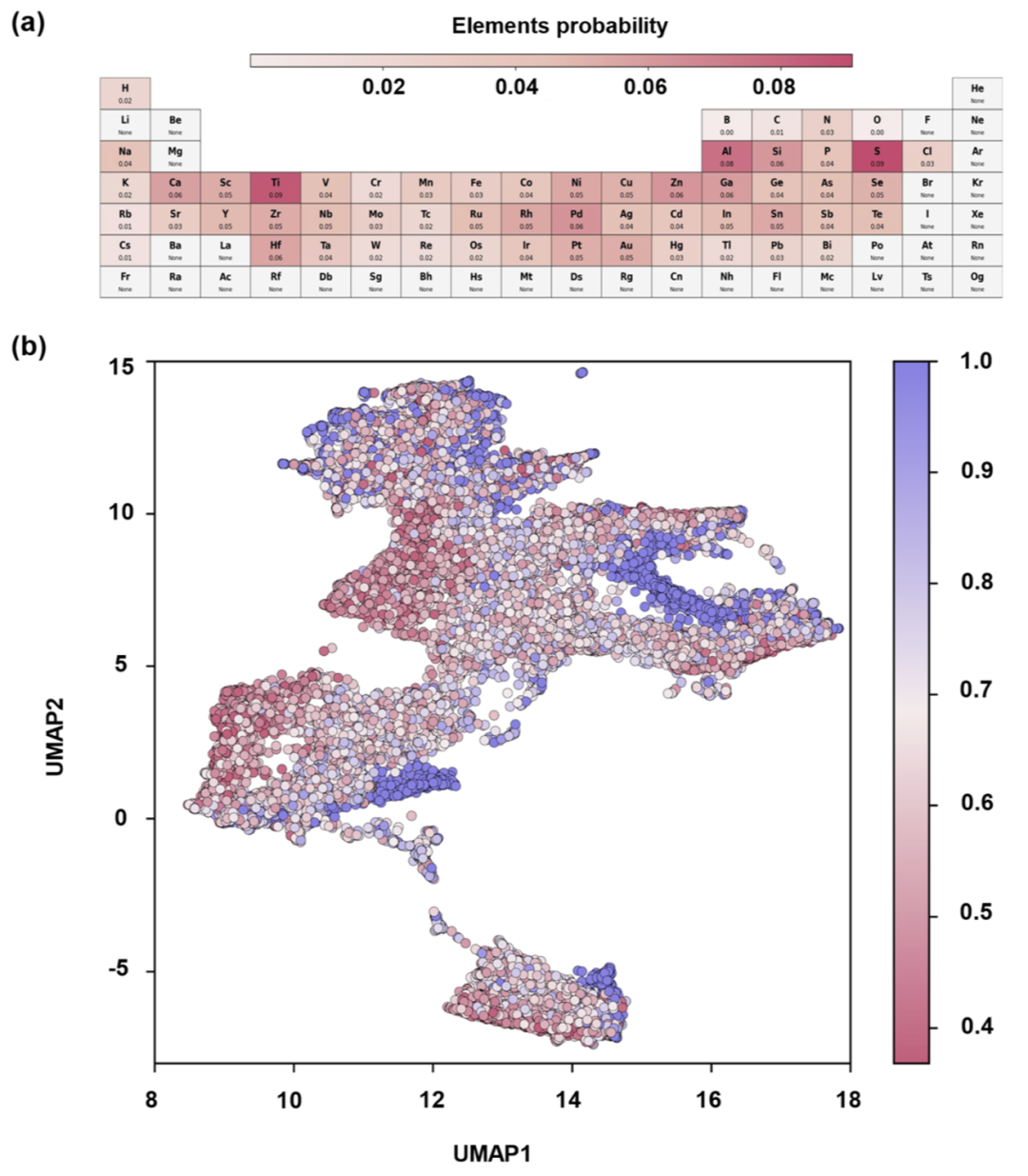
图3:千万级结构的分层筛选流程与 CH3 吸附能分布。
AI重新发现已知甲烷转化催化剂
研究人员随后将结构按元素组成分类为单元、二元和三元催化剂,并统计其平均 CH3 吸附能。结果显示,Ti、Ru、Rh、Ni、Pd 等经典甲烷转化金属均位于最优吸附窗口内。
在二元催化剂分析中,研究人员发现大量实验已知高活性体系,例如 Co–Ru、Ni–Ru、Ni–Rh 等,均落在 −1.5 至 −0.5 eV 的目标窗口区域,并对应低方差区域。这表明框架能够自动重新发现真实有效催化剂。
研究人员进一步统计发现,在恢复出的已知催化剂中,有26种落在目标窗口,而只有少数位于窗口之外。这证明 CH3 吸附能窗口对于催化剂筛选具有较强有效性。
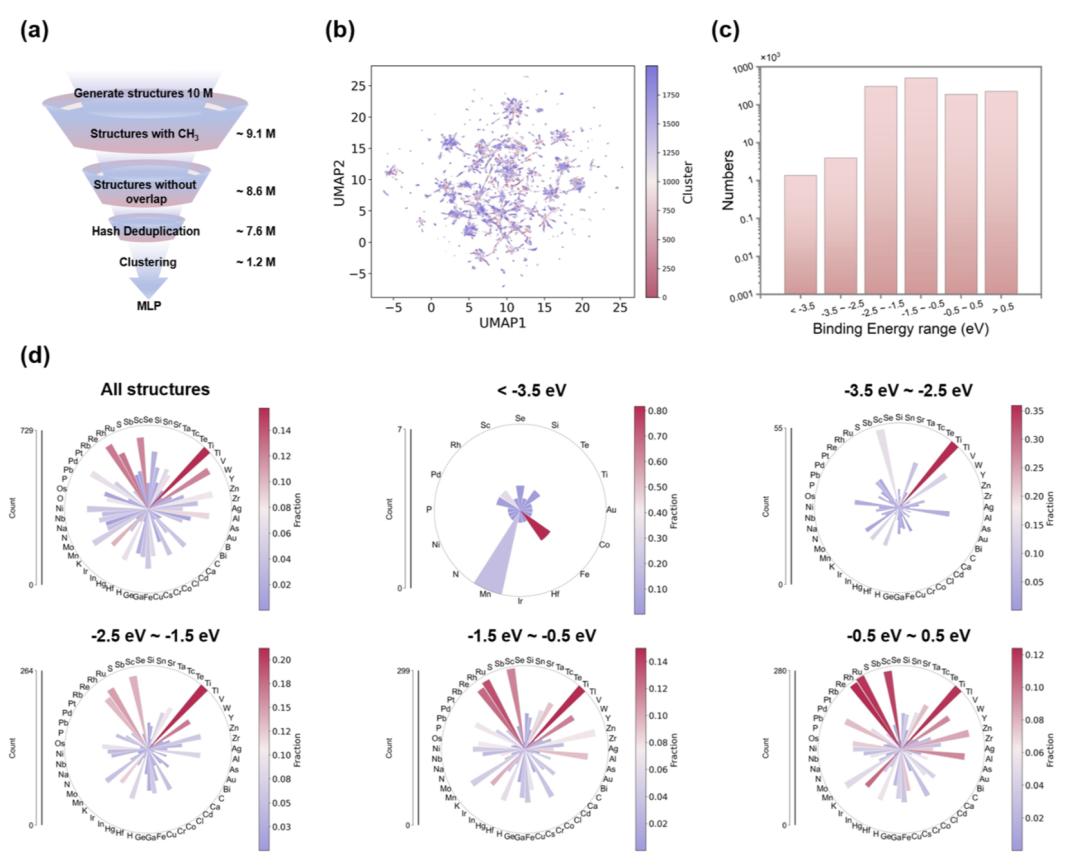
图4:单元、二元与三元催化剂的 CH3 吸附能统计与已知催化剂恢复结果。
SGD分析揭示协同元素调控机制
研究人员进一步利用 subgroup discovery(SGD)方法分析控制 CH3 吸附能的关键描述符。对于二元合金,研究人员发现最佳吸附通常出现在“重过渡金属 + 低价调控金属”的组合中。
例如,Fe、Co、Ni 等具有部分填充 d 轨道的元素能够提供活性位点,而 Al 等低价元素则用于调节表面电子结构。通过 SGD 规则,模型成功恢复了 Co–Mo、Co–Ru、Ni–Rh 等已知有效催化剂。
在三元体系中,研究人员进一步发现一种协同设计模式:一个元素负责调节表面电子结构,而另外两个元素共同调控 d 带填充与带宽。基于这一规律,模型提出了 Al–Ru–Ta、Al–Ni–Ti 与 Mo–Ru–V 等潜在新候选体系。
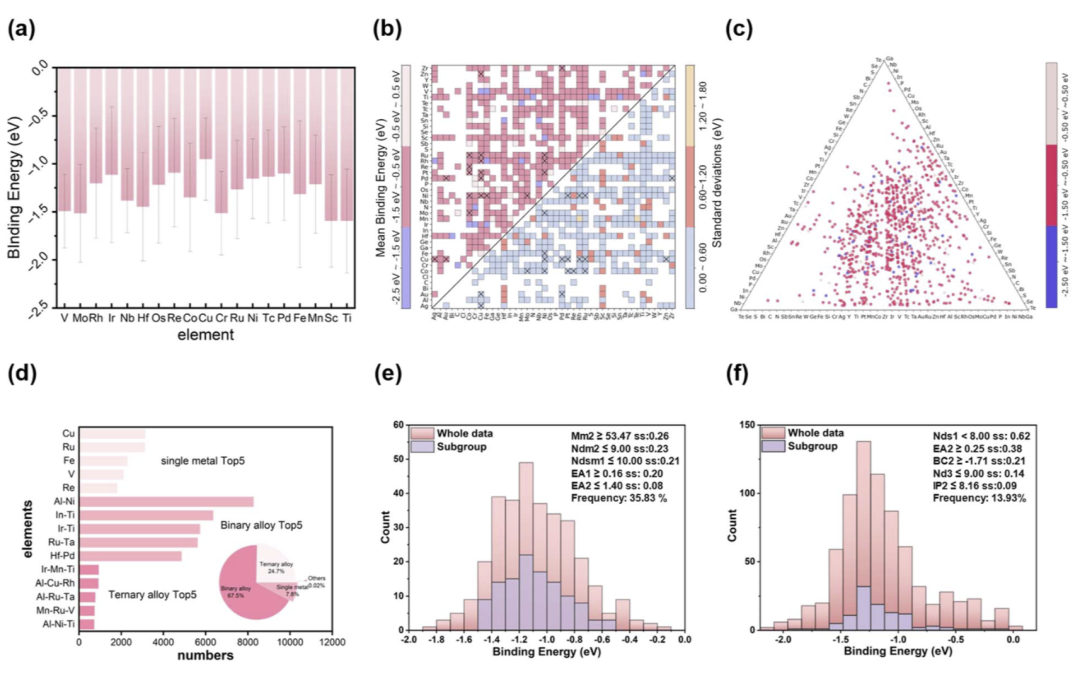
图5:SGD 分析揭示 CH3 吸附能调控的关键元素协同机制。
发现超过1200种未报道甲烷转化候选催化剂
在完整筛选流程后,研究人员最终获得超过1200种此前未报道、但具有潜在甲烷转化活性的候选体系。为了进一步验证结果合理性,研究人员对部分体系进行了 DFT 计算。
结果显示,Ag–Pt 与 Au–Ir–Y 等体系在 CH4 第一阶段 C–H 键断裂过程中表现出较低反应能垒。其中 Ag–Pt 展现出特别低的初始活化能垒,表明其可能成为具有潜力的新型甲烷活化催化剂。
研究人员指出,这些生成结构和预测能量已经全部公开,可作为后续实验验证和催化设计的重要资源。
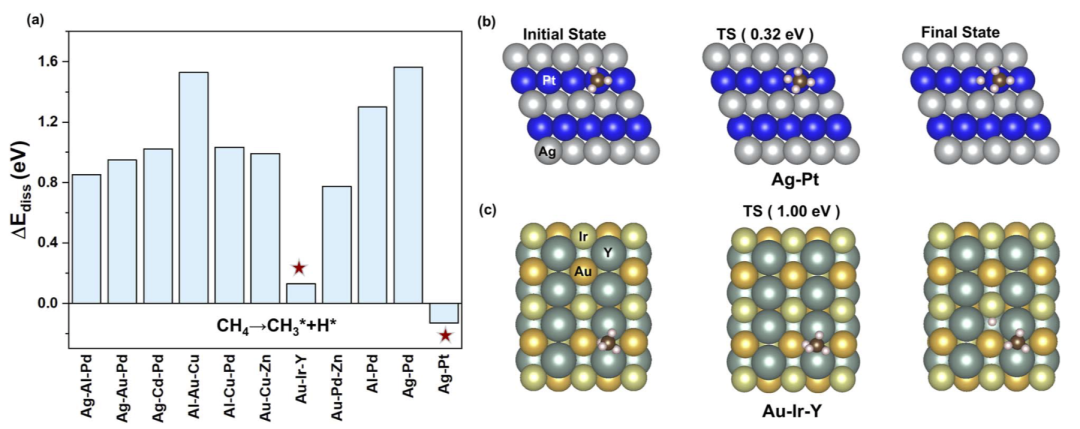
图6:代表性新型甲烷转化催化剂的 DFT 验证与反应能垒分析。
讨论
研究人员构建的分布式生成式 Transformer 框架,首次实现了千万级催化剂结构空间的系统探索。相比已有生成模型,该框架将探索能力提升了两个数量级,并将生成、降维、性能预测与高通量筛选统一到一个可扩展闭环中。
研究结果表明,生成式AI不仅能够重新发现经典催化剂,还能够提出大量此前未知的候选体系。此外,SGD 分析进一步揭示了元素协同调控 CH3 吸附能的机制,为后续催化剂设计提供了可解释性规则。
研究人员认为,该框架未来不仅适用于甲烷转化,还能够推广至其他反应体系与吸附中间体,为大规模催化剂逆向设计与 AI 驱动材料发现提供通用平台。
整理 | DrugOne团队
参考资料
Li, Ruili, et al. "Generative Intelligence Explores Chemical Space at Ten Million Catalysts." Chemical Science (2026).
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-26,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号