Sci. Adv. | AI解码有机分子指纹:定向设计目标能量二维钙钛矿

Sci. Adv. | AI解码有机分子指纹:定向设计目标能量二维钙钛矿

DrugAI
发布于 2026-05-29 13:34:24
发布于 2026-05-29 13:34:24
DRUGONE
人工智能辅助材料发现正在快速改变功能材料的开发模式,但二维杂化钙钛矿的设计仍高度依赖经验与试错法。研究人员提出了一种面向 Dion–Jacobson(DJ)型二维杂化钙钛矿的逆向设计框架,通过构建可逆(invertible)的12维有机分子指纹表示,实现对数百万有机间隔阳离子化学空间的系统探索。
研究人员结合高通量密度泛函理论(DFT)计算、可解释机器学习以及合成可行性筛选,成功识别出能够实现特定能级排列(energy level alignment)的有机分子候选。通过机器学习模型,研究人员不仅能够快速预测 HOMO/LUMO 能级,还揭示了分子结构与二维钙钛矿能级匹配之间的规律。最终,研究人员获得了一系列具有 Ib、IIa 与 IIb 型能级排列的候选 DJ 钙钛矿结构。该研究表明,将具有物理意义且可逆的分子表示融入 AI 材料设计流程,可以显著提高目标性质驱动型杂化材料发现效率,为二维钙钛矿逆向设计提供了新范式。
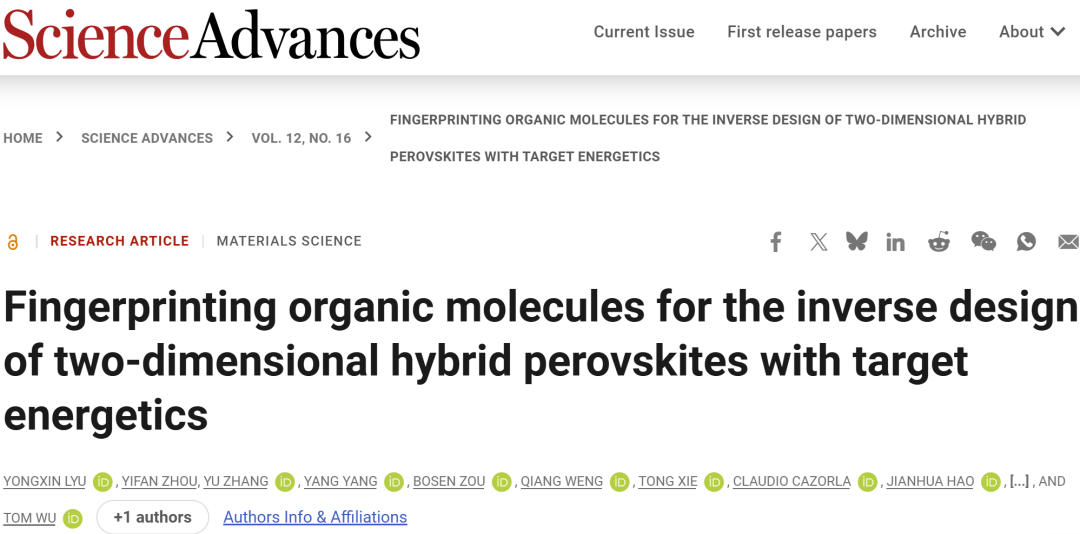
近年来,人工智能与机器学习正在推动材料科学从传统“试错法”向数据驱动设计转变。通过学习已有材料数据中的复杂规律,机器学习模型能够快速预测材料性质、优化结构参数,并实现逆向设计。
二维杂化钙钛矿由于兼具无机半导体与有机分子的特性,在光伏、LED 和光电子器件中展现出巨大潜力。相比三维钙钛矿,二维钙钛矿由于有机阳离子间隔层的存在,拥有更加庞大的设计空间。特别是在 Dion–Jacobson 相结构中,双铵盐有机间隔分子直接连接无机层,不存在范德华间隙,因此能够显著影响载流子输运与能级结构。
然而,目前二维钙钛矿中有机间隔分子的设计主要依赖研究人员经验。例如,通过修改已知分子的官能团,或者借鉴有机光伏中的 π 共轭片段进行尝试。这种方式不仅效率有限,而且无法真正探索庞大的化学空间。
与此同时,能级排列是决定二维钙钛矿性能的关键因素。由于有机层与无机层分别拥有独立能级,它们会形成类似量子阱结构,从而影响电子与空穴在不同层中的分布与传输。然而,目前针对二维钙钛矿能级排列的 AI 辅助设计研究仍十分有限。
因此,研究人员提出构建一种基于可逆分子指纹的逆向设计框架,以实现二维杂化钙钛矿目标能级结构的自动生成与筛选。
方法
研究人员构建了一个针对 DJ 型二维钙钛矿的 AI 辅助逆向设计工作流。首先,研究人员基于已报道的21种双铵盐有机间隔分子,总结其结构规律,并提出一种由12个描述符组成的可逆分子指纹表示方法。该表示能够同时编码芳香环数量、连接方式、氨基位置、杂原子取代、侧链等信息。
随后,研究人员利用“molecular morphing”策略,从基础分子 PDMA 出发,通过13种结构变换操作逐步扩展化学空间,共生成约488万个假想有机间隔分子。研究人员进一步对其中3239种结构进行了高通量 DFT 计算,用于获得二维钙钛矿中的 HOMO/LUMO 与无机层能级排列。
在此基础上,研究人员训练了可解释机器学习模型,用于从分子指纹直接预测 HOMO/LUMO 能级。最后,通过 PubChem 合成可得性筛选以及二维结构 formability score 筛选,对候选分子进行合成可行性评估,并利用 DFT 对最终结构进行验证。
结果
构建可逆分子指纹与二维钙钛矿逆向设计框架
研究人员首先提出了二维 DJ 钙钛矿专用的可逆分子指纹表示。与传统 Morgan fingerprint 等不可逆表示不同,该方法能够从指纹直接反推出一组满足特定性质的候选分子。
研究人员将有机间隔分子拆分为四类结构模块,包括共轭主链、铵盐连接基、杂原子取代以及侧链结构,并进一步编码为12维指纹向量。由于该表示具有较低冗余度与较高可解释性,因此能够高效连接“结构—性质”关系。
研究人员指出,这种可逆性对于逆向设计尤为关键,因为目标不是预测某一个唯一分子,而是生成一组满足目标能级排列的候选结构。
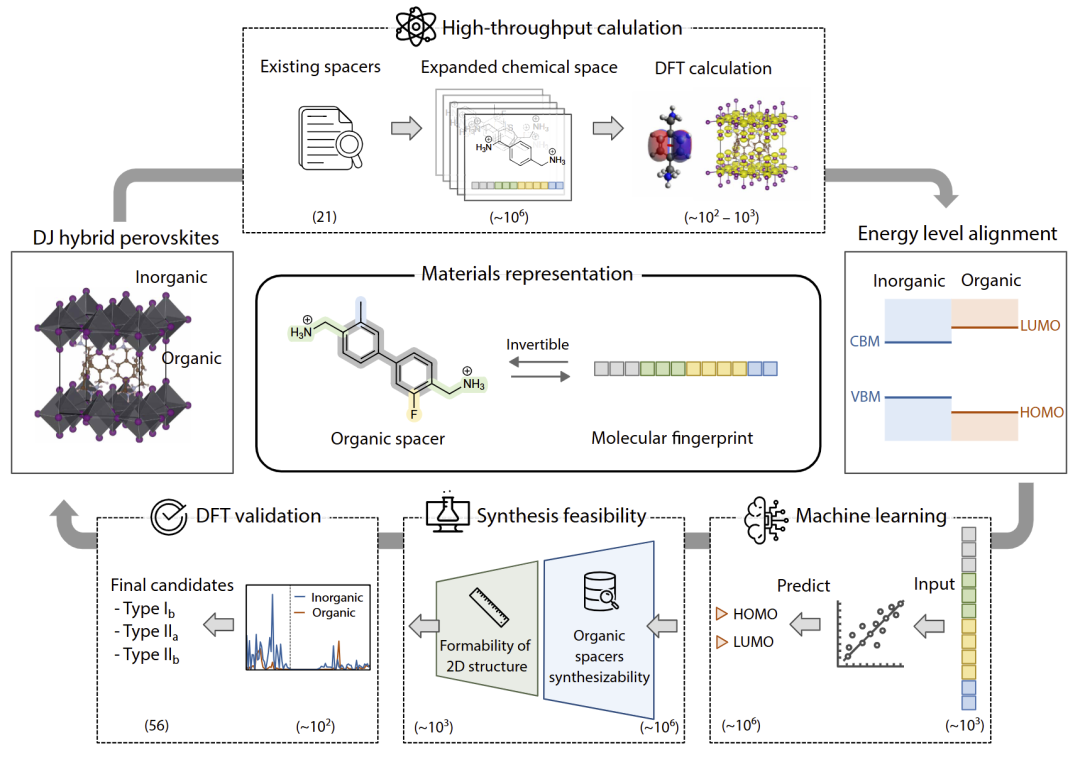
图1:二维 DJ 钙钛矿逆向设计工作流与12维可逆分子指纹表示。
AI生成数百万二维钙钛矿有机间隔分子
研究人员随后使用分子 morphing 方法扩展化学空间。从 Generation 0(PDMA)开始,通过不断引入芳香环、杂原子和侧链修饰,逐步生成更复杂的有机结构。
最终,研究人员从 G0 到 G6 共生成了约488万个假想有机间隔分子,对应21306种不同指纹。所有已知实验有机间隔分子均包含在这一空间中,说明该方法具有较高覆盖度。
研究人员进一步利用 t-SNE 对指纹空间进行降维可视化。结果显示,不同 generation 的分子形成连续而丰富的化学空间分布,高代数结构明显更加复杂。
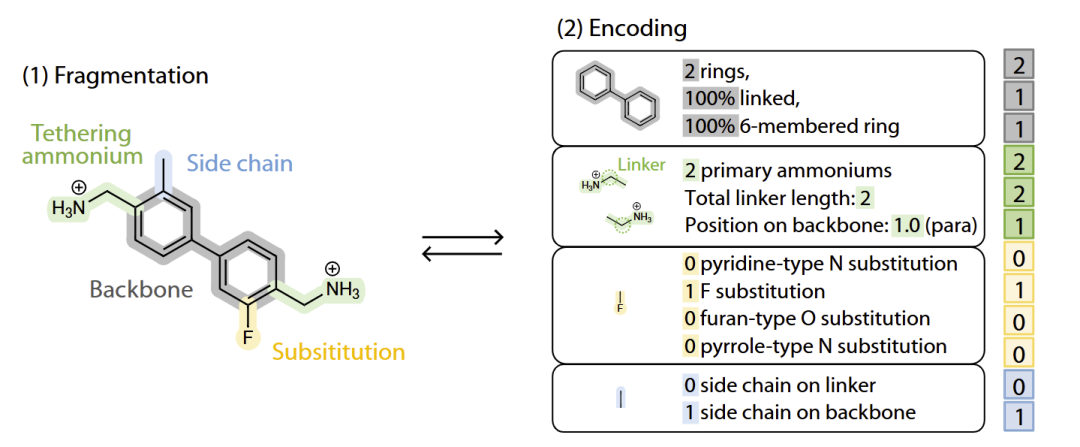
图2:基于 molecular morphing 的百万级有机间隔分子生成与化学空间映射。
高通量DFT揭示二维钙钛矿能级排列规律
研究人员随后对261种 DJ 钙钛矿进行了高通量 DFT 计算,以分析有机层与无机层之间的能级排列关系。
结果显示,大多数已报道二维钙钛矿均属于 type Ia 能级排列,即电子与空穴主要局域于无机层中。相比之下,type IIa 与 IIb 结构则允许有机层参与电子或空穴分布,从而可能产生新的光电性质。
研究人员发现,决定能级排列的主要因素并非无机层,而是有机分子的 HOMO/LUMO 能级。不同有机分子的前线轨道能级跨度可达约6.1 eV,而无机层带边变化仅约0.9 eV。
此外,研究人员还发现,二维钙钛矿中的有机层能级与孤立有机阳离子的 HOMO/LUMO 之间具有强线性关系,因此可以利用单分子计算替代完整晶体计算,大幅降低计算成本。
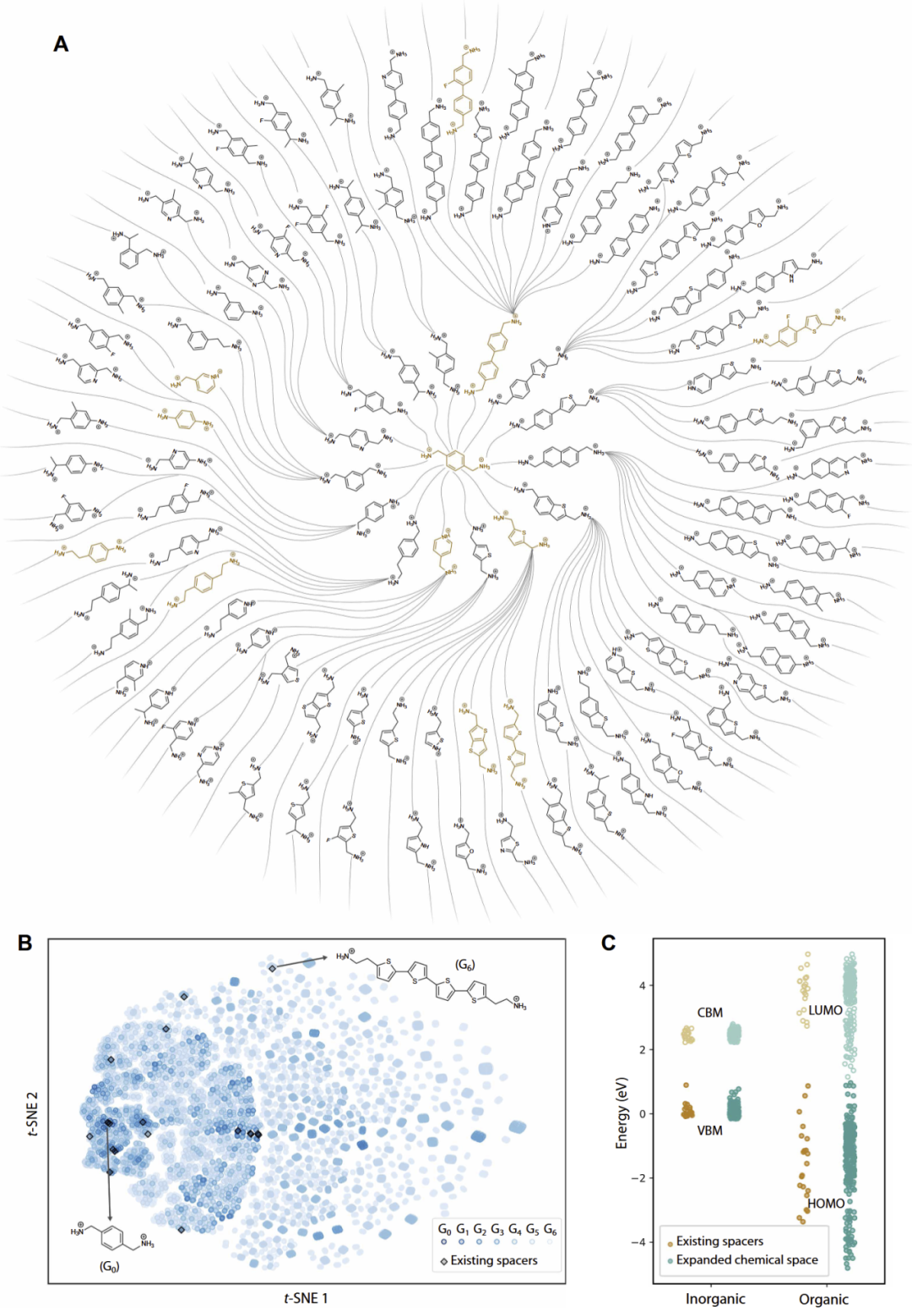
图3:二维 DJ 钙钛矿中的有机-无机能级排列与电子结构分析。
可解释机器学习揭示结构—能级关系
研究人员进一步利用3239个有机间隔分子数据训练机器学习模型,以预测 HOMO/LUMO 能级。
结果显示,无论线性模型还是非线性模型,均能够实现较高预测精度。其中,LASSO 回归由于具备较强可解释性,被用于后续分析。
SHAP 分析表明,影响 HOMO/LUMO 最重要的因素包括:
- 芳香环数量;
- 铵盐连接基数量;
- linker 长度;
- 杂原子类型;
- 主链共轭程度。
研究人员发现:
- 芳香环增加会提高共轭程度,从而提升 HOMO;
- 吡啶型氮原子具有吸电子效应,会降低 HOMO/LUMO;
- 吡咯型氮原子则提高前线轨道能级;
- 氟取代对能级影响相对较弱。
这些规律与有机半导体中的经典设计原则高度一致。
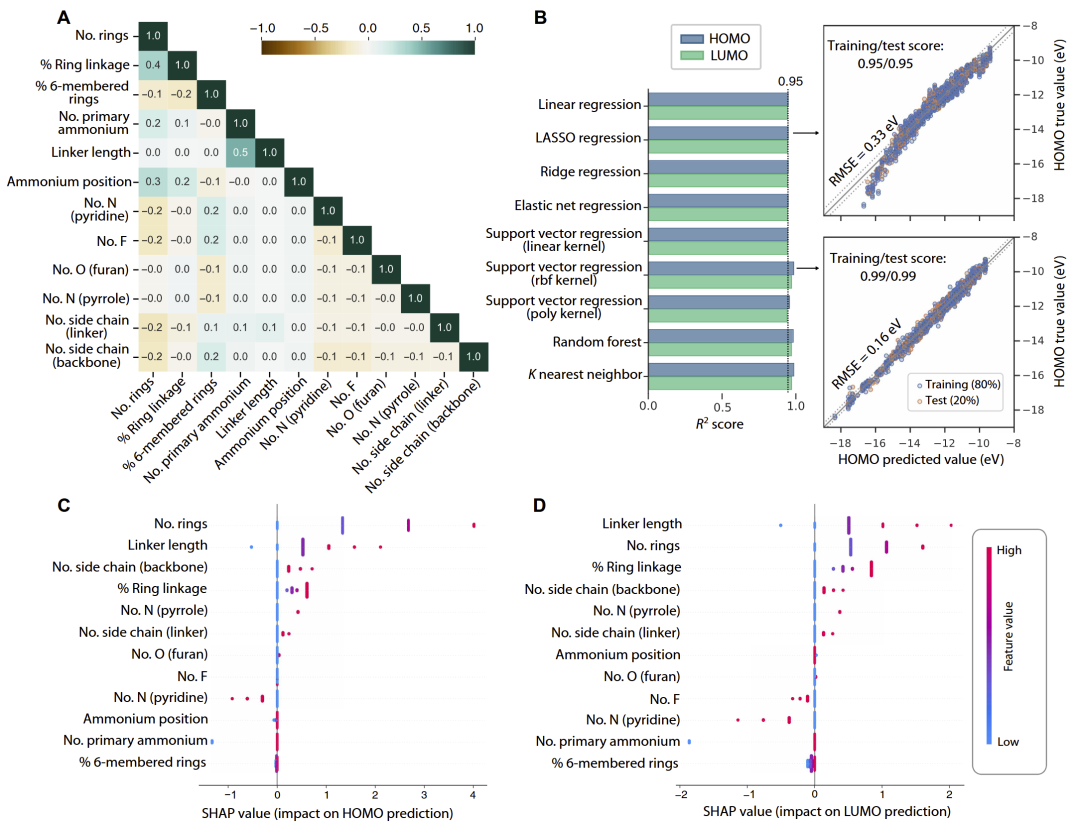
图4:机器学习模型性能与 SHAP 结构—能级关系分析。
构建二维钙钛矿合成可行性筛选体系
研究人员进一步开发了两步式合成可行性筛选框架。
第一步利用 PubChem 数据库评估有机分子的合成可获得性。结果显示,随着 generation 增加,能够在 PubChem 中找到的分子比例逐渐下降,说明复杂结构往往更难合成。
第二步则建立了专门针对 DJ 钙钛矿的 formability score,用于预测某一有机间隔分子是否能够形成二维 DJ 结构。该评分基于五个拓扑描述符,包括:
- 位阻效应;
- N–N 距离;
- 可旋转键数量;
- 分子偏心率;
- 铵基相对位置。
研究人员发现,该 formability score 能够正确区分27/29个已知二维与非二维体系。整体来看,约96.1%的假想分子被预测能够形成 DJ 相结构。
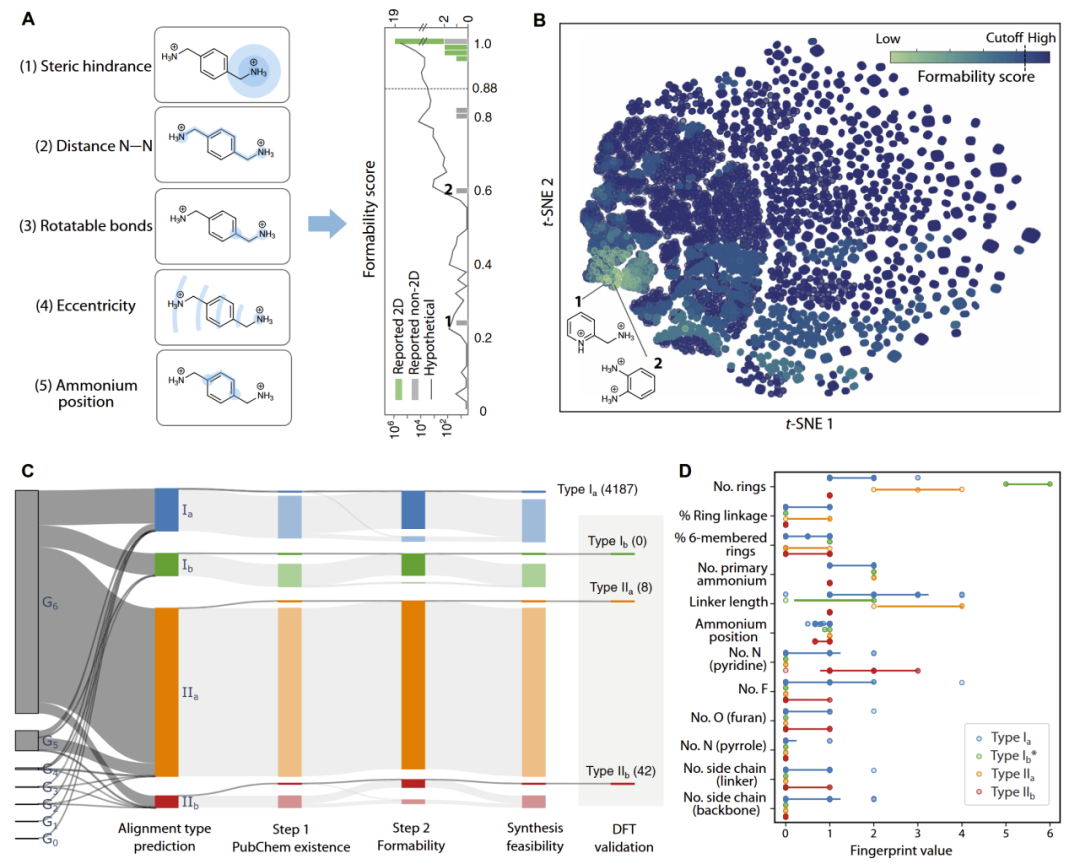
图5:二维 DJ 钙钛矿的 formability score 与合成可行性筛选流程。
AI逆向设计实现目标能级二维钙钛矿发现
在最终逆向设计阶段,研究人员不再穷举整个化学空间,而是直接根据目标能级排列反向约束分子指纹。
结果显示:
- type IIa 分子通常具有更多芳香环与双铵基结构;
- type IIb 分子更倾向于单环与单铵基结构;
- type Ib 则需要极长共轭体系,通常包含5个以上芳香环。
最终,研究人员成功获得:
- 8个 type IIa 候选;
- 44个 type IIb 候选;
- 多个具有潜在 type Ib 能级排列的高共轭候选结构。
这些候选均通过 DFT 验证,并展现出目标导向能级排列特征。
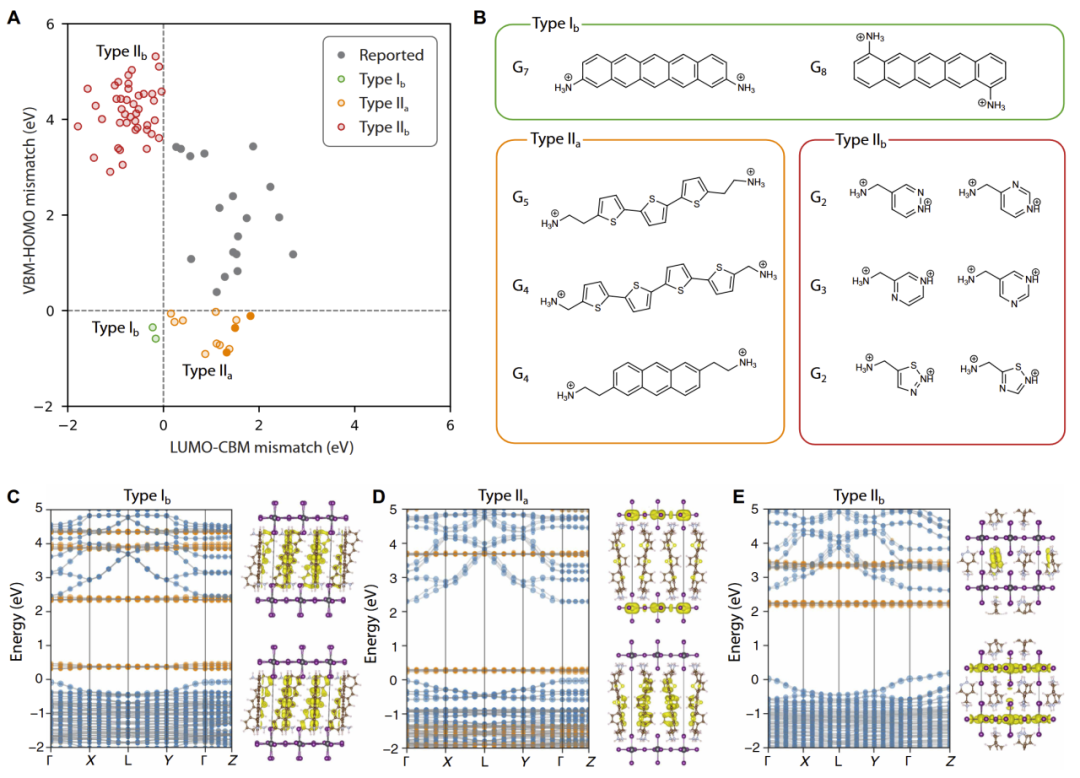
图6:目标能级排列二维 DJ 钙钛矿的逆向设计与最终候选结构。
讨论
研究人员提出了一种面向二维杂化钙钛矿的 AI 辅助逆向设计框架,首次实现了从“目标能级”直接反推有机间隔分子结构。
与传统正向筛选不同,该方法利用可逆分子指纹实现了结构与性质之间的双向映射,使机器学习不仅能够预测性质,还能够反向生成候选分子。研究结果表明,二维钙钛矿中的能级排列主要由有机间隔分子的前线轨道决定,而这一规律能够通过简单、可解释的结构描述符有效捕获。
研究人员认为,该框架未来不仅可用于二维钙钛矿,也有望推广至金属有机框架、杂化半导体及其他有机-无机复合材料体系,推动 AI 驱动的目标性质材料逆向设计。
整理 | DrugOne团队
参考资料
Yongxin Lyu et al. ,Fingerprinting organic molecules for the inverse design of two-dimensional hybrid perovskites with target energetics.Sci. Adv.12,eaeb4144(2026).
DOI:10.1126/sciadv.aeb4144
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-05-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号