Science | 从骨架到序列:ProteinMPNN 如何让蛋白设计更可靠?

Science | 从骨架到序列:ProteinMPNN 如何让蛋白设计更可靠?

DrugIntel
发布于 2026-07-06 17:26:13
发布于 2026-07-06 17:26:13

文献来源: J. Dauparas et al., Robust deep learning–based protein sequence design using ProteinMPNN. Science, 378, 49-56(2022). DOI:10.1126/science.add2187
摘要
蛋白质设计的核心难题之一,是给定一个目标骨架结构,找到能够稳定折叠到该结构的氨基酸序列。传统 Rosetta 方法依赖能量函数与侧链构象搜索,计算成本高,也常需要专家经验。ProteinMPNN 将这一问题转化为基于蛋白骨架图的条件序列生成:用主链原子间距离构建局部几何环境,通过消息传递网络编码骨架,再以随机自回归顺序生成序列。论文不仅在 native sequence recovery 上显著超过 Rosetta,还通过晶体结构、冷冻电镜和功能实验验证了其在单体、寡聚体、纳米颗粒和蛋白结合设计中的实用价值。
1. 为什么这篇论文值得关注?
过去几年,蛋白质结构预测取得了巨大进展,但蛋白质设计并不是结构预测的反问题那么简单。
结构预测回答的是:给定一条天然或人工序列,它可能折叠成什么结构? 蛋白设计要回答的是:给定一个我们想要的结构、界面或功能构型,应该给出怎样的氨基酸序列,让它真的折叠成这个结构,并在实验中表达、稳定、组装或结合目标?
这一步在 AI 蛋白设计中非常关键。因为很多生成模型可以产生骨架,但骨架本身不能表达、不能纯化、不能发挥功能。真正进入实验体系的是序列。没有可靠的序列设计,骨架生成只是图纸;有了可靠的序列设计,图纸才可能变成蛋白。
ProteinMPNN 的意义就在这里:它专注解决固定骨架序列设计问题,即给定蛋白主链坐标,快速生成与该骨架匹配的氨基酸序列。论文显示,在 native protein backbone 上,ProteinMPNN 的序列恢复率为 52.4%,明显高于 Rosetta 的 32.9%;同时,ProteinMPNN 在 100 个残基的设计任务上单 CPU 约 1.2 秒,而 Rosetta 约 258.8 秒。
这不仅是速度提升,更意味着蛋白设计流程可以从少量手工调参,转向大规模候选序列生成、筛选与实验验证。
2. 研究背景:蛋白序列设计为什么重要?
蛋白质设计通常可以拆成两个相互依赖的问题:
第一步是结构设计,即设计一个合理的三维骨架。这个骨架可能是单体蛋白、对称寡聚体、纳米颗粒、蛋白结合剂,或者承载功能基序的 scaffold。
第二步是序列设计,即找到一条氨基酸序列,使它能够稳定折叠到这个目标骨架,同时尽可能满足溶解性、表达量、组装状态和功能需求。
传统 Rosetta 序列设计通常把问题看作能量优化:在固定主链上搜索不同氨基酸类型和侧链 rotamer 组合,寻找低能量构象。这种思路有明确物理基础,但也存在三个现实困难。
第一,搜索空间巨大。每个位置有 20 种氨基酸,侧链还有多个 rotamer,组合空间随残基数指数增长。
第二,能量函数并不完美。蛋白稳定性、溶解性、表达、界面组装与功能选择性并不能被一个静态能量函数完全描述。
第三,实际设计往往需要大量人工经验。尤其是对称复合物、纳米颗粒和 protein-protein interface,设计者通常要反复调节疏水性、界面 packing、表面电荷和构象约束。
ProteinMPNN 的核心判断是:如果目标骨架已经给定,那么某个位置应该选择什么氨基酸,主要由其局部三维几何环境决定。换句话说,蛋白序列设计可以被看作结构到序列的条件概率建模问题,而不必每次都显式枚举所有侧链构象。
3. 这篇论文的核心思想
ProteinMPNN 的核心思想可以概括为一句话:
把固定骨架蛋白设计看作一个几何条件下的自回归序列生成问题。
输入不是序列,而是蛋白主链结构。模型先把骨架转化为残基图:每个残基是一个节点,残基之间根据空间邻近关系建立边;边特征主要来自 N、Cα、C、O 和虚拟 Cβ 等主链相关原子之间的距离。然后,消息传递神经网络在这个图上聚合局部几何信息,最后按照某种解码顺序逐步生成氨基酸序列。
这套设计有几个关键点:
第一,它不直接做侧链 rotamer 搜索,而是学习结构环境到氨基酸分布的映射。
第二,它使用随机解码顺序,而不是固定从 N 端到 C 端生成序列,因此可以自然支持固定残基、固定基序、局部重设计和 binder 设计。
第三,它允许把不同位置的氨基酸概率绑定在一起,从而支持同源多聚体、重复蛋白、对称设计和多状态设计。
第四,它通过在训练中加入主链噪声,提高对非理想骨架的鲁棒性。这一点很重要,因为真实设计骨架往往来自 Rosetta、AlphaFold hallucination 或扩散模型,并不具备天然晶体结构那样的高精度。
5. 方法细节:ProteinMPNN 到底是怎么做的?
5.1 任务定义与输入输出
ProteinMPNN 解决的是 fixed-backbone protein sequence design。
输入: 一个目标蛋白骨架结构,包括主链原子坐标。论文重点使用 N、Cα、C、O 以及基于主链几何放置的虚拟 Cβ 原子。
输出: 与目标骨架匹配的氨基酸序列。对于多链复合物,输出可以包含每条链的序列;对于对称设计,某些位置可以被约束为相同氨基酸;对于 binder 或功能基序设计,部分位置可以固定不变。
这意味着 ProteinMPNN 本身不是骨架生成模型。它不负责创造蛋白主链,而是给已有骨架配上序列。因此,它特别适合作为 RFdiffusion、Rosetta backbone design、AlphaFold hallucination 等结构生成方法之后的序列设计模块。
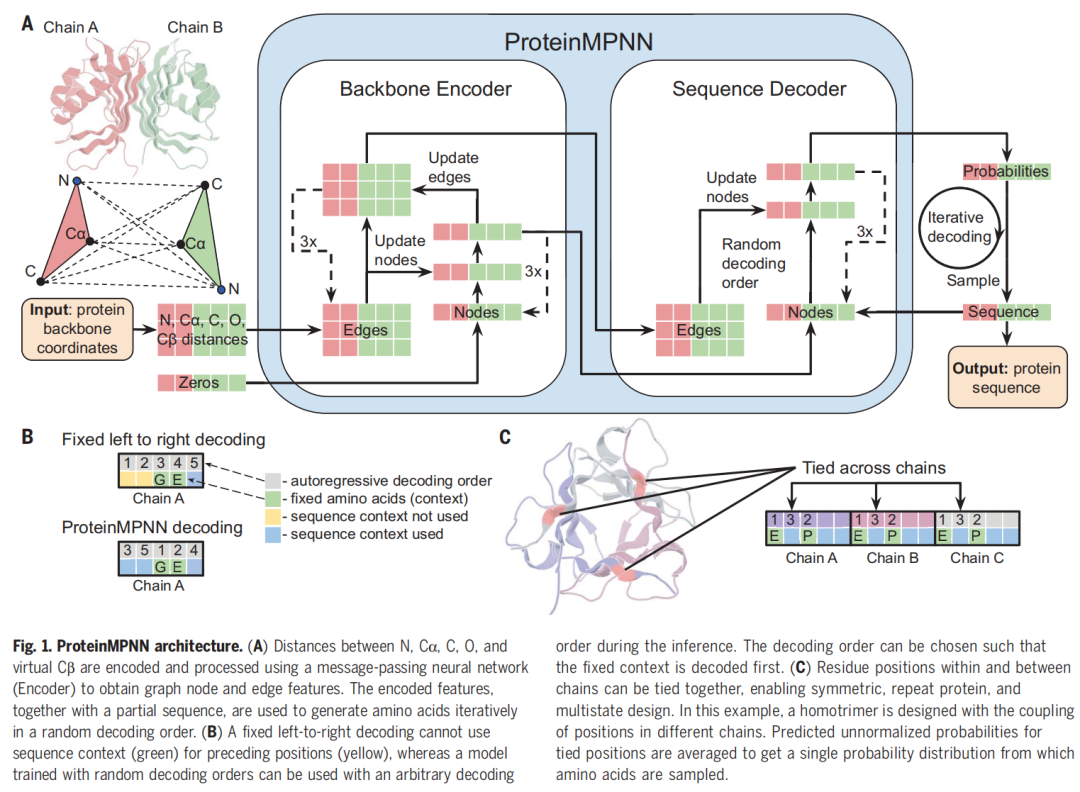
5.2 数据表示:把蛋白骨架变成几何图
ProteinMPNN 的输入表示非常关键。
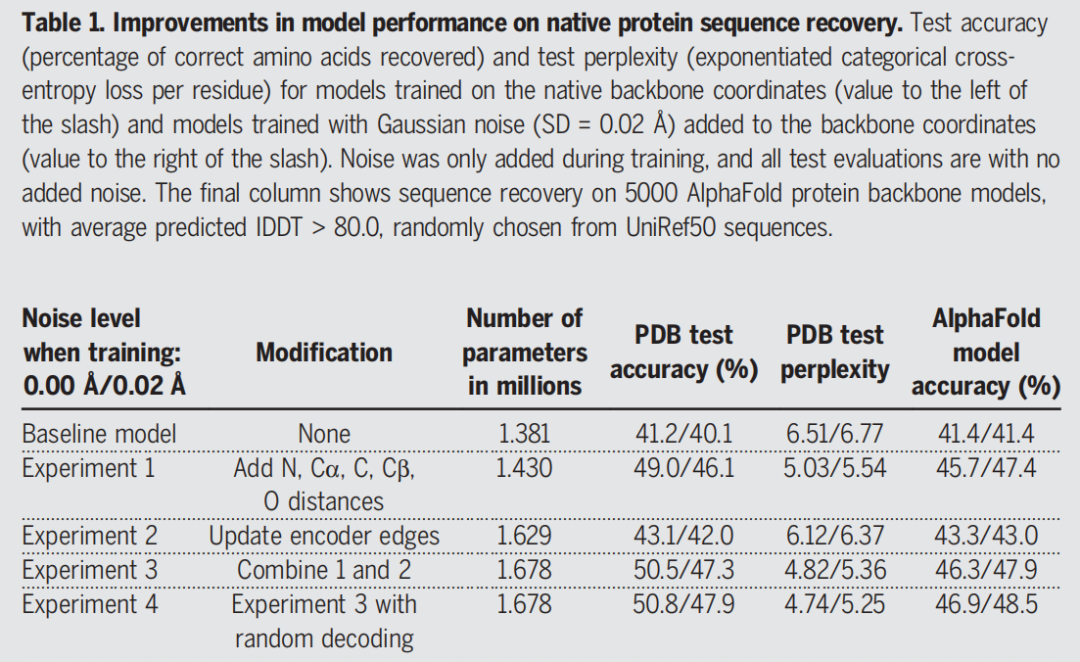
论文从一个已有 MPNN 模型出发,原始特征包括 Cα-Cα 距离、局部坐标框架方向和主链二面角。作者发现,加入 N、Cα、C、O、虚拟 Cβ 之间的距离后,序列恢复率从 baseline 的 41.2% 提升到 49.0%。这说明对于结构到序列映射,原子间距离比单纯的二面角或框架方向提供了更直接的几何归纳偏置。
可以这样理解:氨基酸选择不是只由一个残基自身的二级结构决定,而是由周围空间环境共同决定。一个疏水核心位置可能偏好 Leu、Ile、Val、Phe 等;一个暴露表面位置可能更偏好极性或带电残基;一个界面位置则取决于另一条链上的几何互补和相互作用环境。
ProteinMPNN 用局部邻域图捕捉这种信息。作者测试了 16、24、32、48、64 个最近 Cα 邻居,发现性能在 32 到 48 个邻居附近趋于饱和。这个结果很有启发性:序列设计比结构预测更局部,某个位置的氨基酸身份主要由附近结构环境决定,而不是必须依赖全局长程信息。
5.3 模型结构:骨架编码器与序列解码器
ProteinMPNN 的整体结构包含两个核心部分:Backbone Encoder 和 Sequence Decoder。
第一部分:Backbone Encoder。 输入蛋白骨架图后,编码器通过消息传递更新节点和边特征。节点对应残基,边对应残基之间的空间关系。作者发现,在编码器中不仅更新节点,也更新边,可以进一步提升性能;将原子间距离特征和边更新结合后,PDB 测试集序列恢复率达到 50.5%。
第二部分:Sequence Decoder。 解码器在编码后的结构特征基础上,逐步预测每个位置的氨基酸概率。它是自回归模型,也就是说,已经生成或固定的序列位置会作为上下文,影响后续位置的氨基酸选择。
论文图 1 展示了这一流程:主链坐标进入模型,先转化为 N、Cα、C、O、虚拟 Cβ 的距离特征,再经过 MPNN 编码器生成节点与边表征,随后解码器结合部分序列上下文,按随机顺序迭代生成氨基酸概率并采样序列。
5.4 为什么随机解码顺序很重要?
如果模型只能从 N 端到 C 端生成序列,它在很多设计场景中会很不方便。
例如,设计 protein binder 时,目标蛋白序列通常已经给定,或者某个功能基序必须固定。固定从左到右解码时,模型不能灵活利用这些已知区域作为上下文。
ProteinMPNN 改用 order-agnostic autoregressive model:训练时随机采样解码顺序,推理时可以指定任意解码顺序。这样,固定残基或固定基序可以先作为上下文提供给模型,然后模型只设计其余位置。论文指出,这种随机解码不仅带来小幅序列恢复率提升,还让模型能够用于更广泛的单链和多链设计任务。
这一步是 ProteinMPNN 从 benchmark 模型走向实用工具的关键。它让模型不只是会给完整单体骨架设计序列,也能处理真实蛋白工程中常见的约束设计问题。
5.5 对称、多链和多状态设计如何实现?
ProteinMPNN 的另一个重要设计是 tied positions。
对于同源二聚体或三聚体,链 A 的第 i 个位置和链 B 的第 i 个位置往往需要使用相同氨基酸。ProteinMPNN 的做法是:先分别预测这些等价位置的未归一化概率,再把这些概率合并,形成一个共同的氨基酸分布,然后从中采样同一个氨基酸。
这使它可以自然处理:
- • 同源多聚体设计;
- • 循环对称蛋白设计;
- • 重复蛋白设计;
- • 纳米颗粒中跨链等价位点设计;
- • 多状态设计中的正设计与负设计。
多状态设计尤其值得注意。论文提到,可以对不同 backbone state 的未归一化概率进行线性组合,用正系数强化希望稳定的状态,用负系数削弱不希望出现的状态。虽然这一点在论文中不是最主要的实验重点,但它提示 ProteinMPNN 不只是单状态序列生成器,也可以作为更复杂设计约束的概率模块。
5.6 训练目标与噪声训练
ProteinMPNN 的训练目标本质上是预测给定结构条件下的氨基酸类别,因此使用类别交叉熵,对应论文中报告的 perplexity 和 sequence recovery。
训练数据方面,作者最初使用 19,700 个 PDB 高分辨率单链结构,并按 CATH 分类进行 80/10/10 的训练、验证和测试划分;最终模型则在 PDB 蛋白组装体上训练,纳入截至 2021 年 8 月 2 日、由 X-ray 或 cryo-EM 解析、分辨率优于 3.5 Å 且残基数少于 10,000 的结构。
噪声训练是论文中非常有价值的一点。作者发现,在主链坐标中加入小的高斯噪声,会降低在精确 PDB backbone 上的序列恢复率,却能提高模型在 AlphaFold 预测骨架上的表现。原因可能是,晶体结构中包含一些与天然氨基酸身份相关的精细几何痕迹,模型如果过度拟合这些痕迹,反而不利于真实设计场景。加入噪声后,模型更关注整体拓扑和极性/疏水模式,而不是局部坐标细节。
这对实际设计很重要。因为 de novo backbone 不一定是原子级精准结构,模型必须对小几何误差具有鲁棒性。
5.7 推理、采样与实际使用流程
ProteinMPNN 推理时,用户输入一个目标 backbone,可以选择固定部分残基、绑定对称位置、设定采样温度,然后生成多个候选序列。
采样温度控制多样性。论文显示,提高温度可以显著增加序列多样性,同时只带来较小的平均序列恢复率下降。模型还可以用平均 log probability 作为序列质量指标,用于快速排序和筛选候选序列。
一个实际使用流程大致如下:
- 1. 使用 Rosetta、RFdiffusion、AlphaFold hallucination 或其他方法生成目标骨架;
- 2. 将骨架坐标输入 ProteinMPNN;
- 3. 指定固定基序、固定目标链或对称绑定位置;
- 4. 采样生成多条候选序列;
- 5. 用 AlphaFold 或 RoseTTAFold 检查单序列是否能回折到目标结构;
- 6. 进一步根据表达、溶解性、界面、功能位点和实验成本筛选;
- 7. 合成基因并进行表达、纯化、结构或功能验证。
这也是为什么 ProteinMPNN 后来会成为 AI 蛋白设计工作流中的基础模块:它速度快、约束灵活、能批量生成候选序列,而且容易与结构预测模型和实验筛选闭环结合。
6. 实验设计与关键结果
6.1 In silico 评估:不只是高一点的序列恢复率
论文首先比较 ProteinMPNN 与 Rosetta fixed-backbone sequence design。结果显示,在 402 个单体 backbone 测试中,ProteinMPNN 的 native sequence recovery 为 52.4%,Rosetta 为 32.9%;同时 ProteinMPNN 在不同埋藏程度的残基上都优于 Rosetta。
这里的 sequence recovery 衡量的是模型在给定天然骨架时恢复天然氨基酸的比例。它不是蛋白设计成功率,但可以反映模型是否学到了结构环境与氨基酸偏好之间的统计关系。
作者进一步在 690 个单体、732 个同源多聚体和 98 个异源多聚体上测试,median sequence recovery 分别为 52%、55% 和 51%;界面残基的恢复率在同源和异源复合物中也分别达到 53% 和 51%。这说明 ProteinMPNN 不局限于单链蛋白,也能处理界面环境。
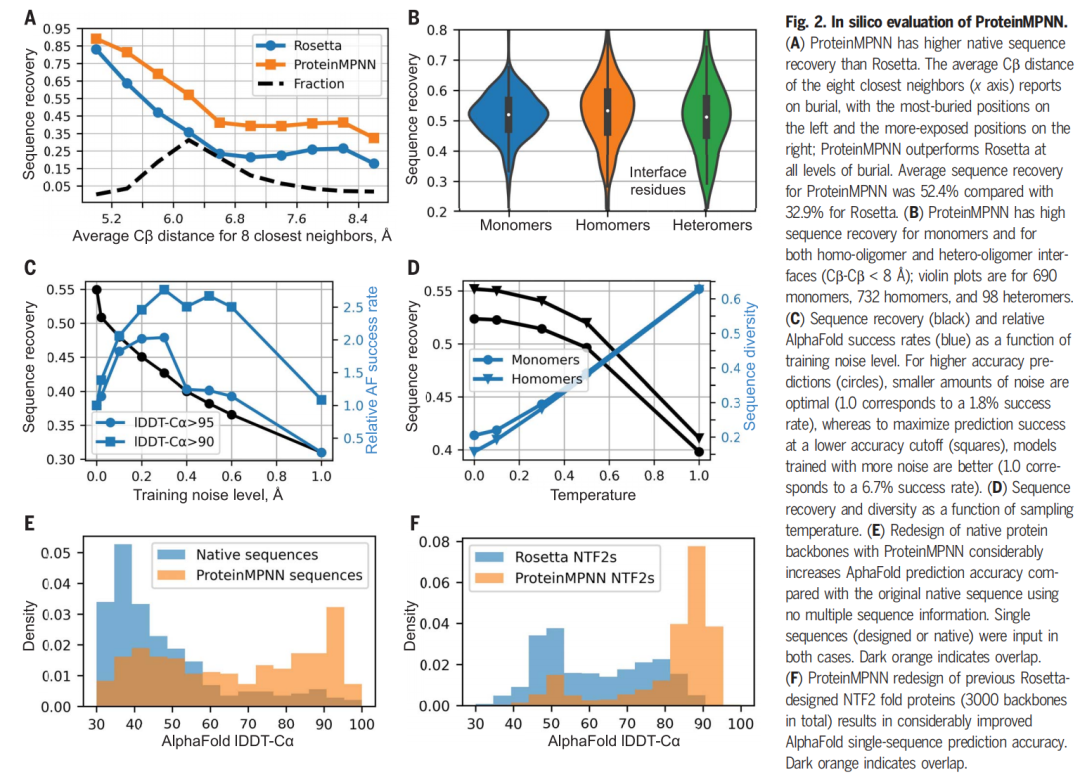
值得注意的是,深层核心位置的恢复率可达 90–95%,而表面位置约 35%。这符合蛋白生物物理直觉:核心区域几何约束更强,氨基酸选择更确定;表面区域可接受更多序列变化,因此天然序列不一定是唯一合理答案。
6.2 AlphaFold 评估:设计序列是否更能编码目标结构?
论文进一步用 AlphaFold 检查 ProteinMPNN 生成序列是否能更强地编码目标结构。
一个有代表性的结果是:对于 Rosetta 生成的含小分子结合口袋的 de novo scaffold,原始设计序列中只有 2.7% 被 AlphaFold 预测能折叠到目标结构;经过 ProteinMPNN 重新设计后,这一比例提升到 54.1%。
这说明 ProteinMPNN 不只是复现天然序列,也能提高人工设计骨架的 sequence-to-structure 一致性。对酶设计、小分子结合蛋白设计和功能 scaffold 设计而言,这一点非常关键。
6.3 实验验证:这篇论文最有分量的部分
ProteinMPNN 论文最值得重视的地方,是它不止停留在计算评估,而是进行了系统的实验验证。
AlphaFold hallucination 设计的救援
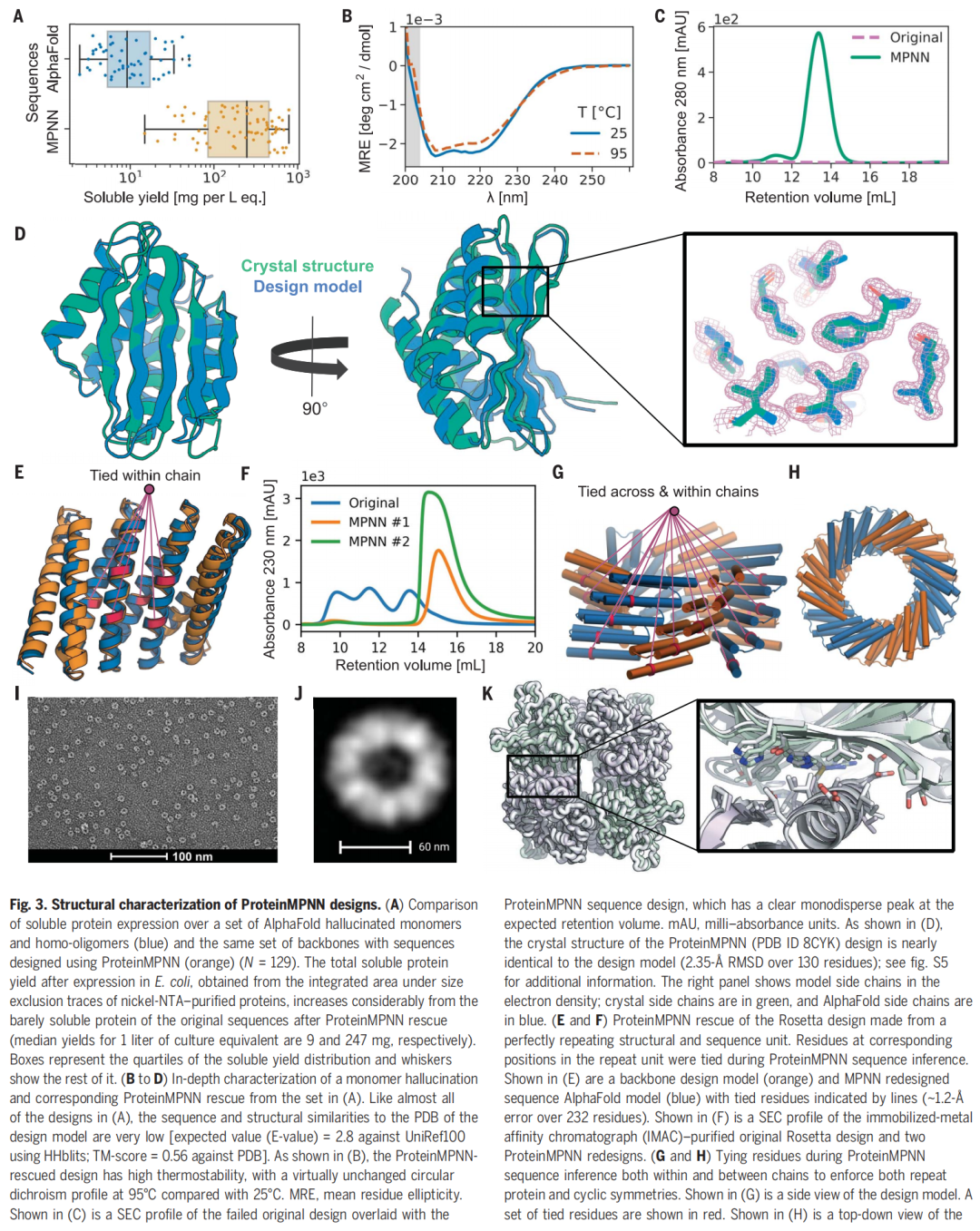
作者首先测试 AlphaFold hallucination 生成的单体和同源寡聚体。原始 hallucinated sequences 多数不溶,median soluble yield 只有 9 mg/L culture equivalent。ProteinMPNN 重新设计同一批 backbone 后,在 96 个尝试表达的设计中,73 个可溶表达,50 个通过 SEC 显示目标单体或寡聚状态,median soluble yield 提升到 247 mg/L culture equivalent。
其中一个单体设计的晶体结构与设计模型非常接近,130 个残基上的 RMSD 为 2.35 Å,并且圆二色谱显示在 95°C 下仍保持二级结构特征。
重复蛋白、循环对称蛋白与纳米颗粒
作者利用 tied positions 设计重复蛋白和对称寡聚体。对于 C5/C6 循环寡聚体,Rosetta 设计组 10 个中只有 4 个可溶,且没有一个通过 SEC-MALS 确认为正确寡聚状态;ProteinMPNN 设计组 18 个中有 16 个可溶,5 个具有正确寡聚状态。
在四面体蛋白纳米颗粒设计中,ProteinMPNN 对 27 个 backbone 设计了 76 条序列,不需要额外人工干预;其中 13 个形成约 1 MDa 的预期组装体,并且一个晶体结构与设计模型非常接近,两个亚基上的 Cα RMSD 为 1.2 Å。
蛋白功能设计:SH3 binder
最后,作者测试了一个更难的问题:设计能展示 polyproline II helix motif 并结合 Grb2 SH3 domain 的蛋白。作者固定核心 SH3-binding motif PPPRPPK,用 ProteinMPNN 重新设计 scaffold 序列。BLI 实验显示,设计蛋白对 Grb2 SH3 具有强结合信号,且破坏关键相互作用的点突变会消除结合。
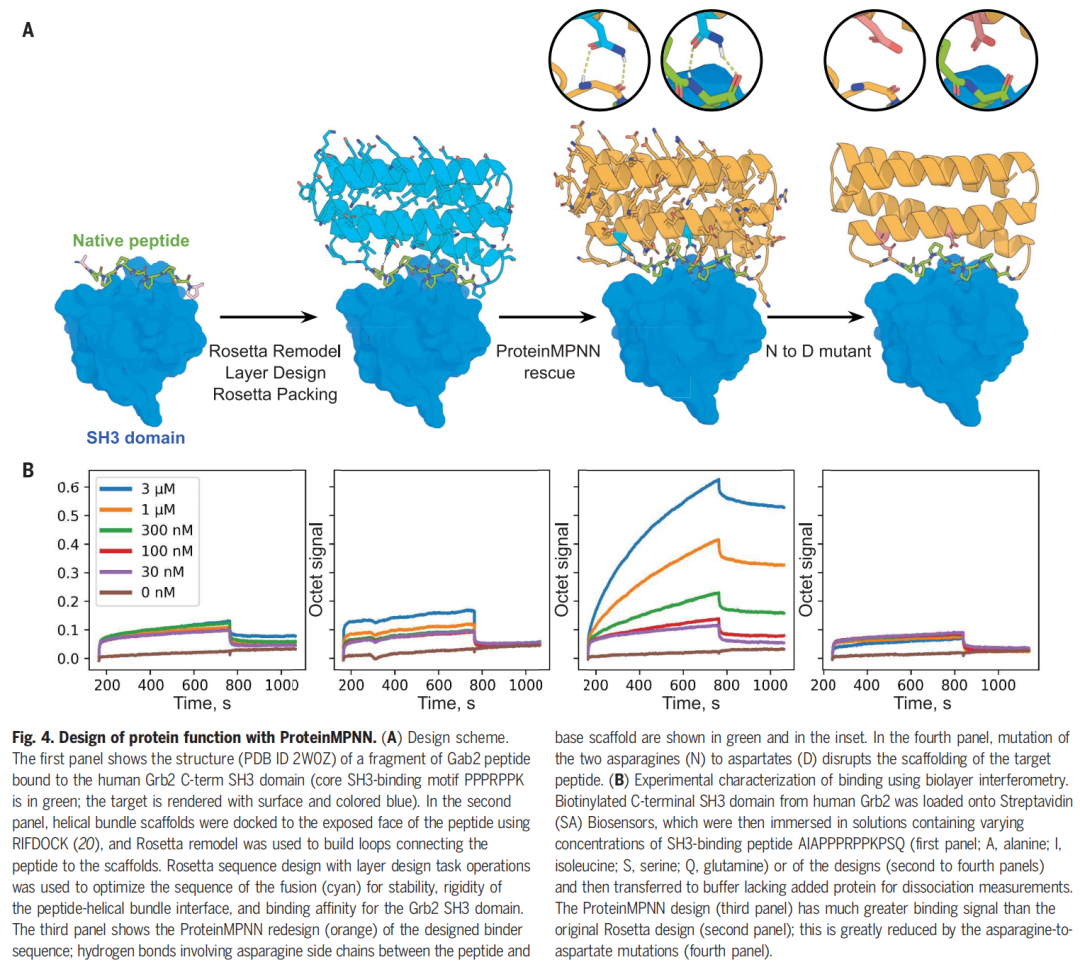
这一结果说明 ProteinMPNN 可以在固定功能基序的前提下设计周围 scaffold,使功能基序被正确呈现并产生目标结合。
7. 这篇文章的真正启发
启发一:蛋白设计中的关键瓶颈不只是生成骨架,而是让序列编码骨架
很多 AI 蛋白设计讨论容易聚焦 backbone generation,但真正决定实验可行性的,是序列能否稳定编码该骨架。ProteinMPNN 让结构生成和序列设计之间有了一个高效接口。
启发二:局部几何足以支撑高质量序列设计
ProteinMPNN 的近邻图结果提示,对固定骨架序列设计而言,局部环境具有强决定性。这与结构预测需要长程共进化信息不同,也解释了为什么较小的 MPNN 模型就能表现很好。
启发三:约束生成比无约束生成更接近真实蛋白工程
随机解码、固定残基、绑定等价位置、多链处理,这些功能看起来不像花哨模型创新,但正是它们让 ProteinMPNN 能处理真实设计任务。真实蛋白工程往往不是从零生成,而是在约束下寻找可行序列。
启发四:实验验证决定蛋白设计方法的可信度
ProteinMPNN 的影响力不只来自 sequence recovery,而来自 X-ray、cryo-EM、SEC、SEC-MALS、CD 和 BLI 等实验验证。对蛋白设计来说,计算指标只能筛选候选,不能替代最终实验。
8. 局限性与值得警惕的问题
ProteinMPNN 很强,但不能把它理解为蛋白设计的完整解决方案。
第一,它依赖输入骨架质量。如果 backbone 本身不可折叠、几何不合理、功能位点摆放错误,ProteinMPNN 很难从序列层面彻底拯救。
第二,它主要解决固定骨架序列设计,不直接优化蛋白动力学、构象变化、表达宿主依赖性、免疫原性或长期稳定性。
第三,sequence recovery 不是设计成功率。天然序列恢复率高,说明模型学到了天然蛋白统计规律,但 de novo design 的目标通常不是恢复天然序列,而是产生能表达、折叠和发挥功能的新序列。
第四,AlphaFold 过滤会带来方法学偏倚。用 AlphaFold 判断设计序列是否回折到目标结构非常实用,但也可能让设计流程偏向 AlphaFold 容易预测的序列模式,而不是所有真实可折叠序列。
第五,功能设计仍然需要额外建模。ProteinMPNN 可以保留功能基序、设计 scaffold,但并不等同于直接优化催化活性、结合自由能、选择性或细胞环境中的功能表现。
因此,ProteinMPNN 更准确的定位是:一个高效、通用、实验表现很强的结构条件序列设计器,而不是单独完成蛋白功能设计的全流程系统。
9. 对 AI 制药和蛋白设计未来的意义
从 AI 制药角度看,ProteinMPNN 的意义不只在蛋白质工程,也在于它为结构生成模型提供了一个可落地的序列化接口。
在当前 AI 蛋白设计流程中,一个典型组合是:
RFdiffusion 或其他模型生成目标骨架 → ProteinMPNN 设计序列 → AlphaFold/RoseTTAFold 回折验证 → Rosetta 或物理方法精修 → 实验表达与功能测试。
这套流程背后的逻辑是分工:生成模型负责探索结构空间,ProteinMPNN 负责把结构转译为序列,结构预测模型负责快速筛选,实验负责最终判定。
对药物发现而言,这种能力可以用于多个方向:
- • 设计蛋白 binder,用于阻断蛋白-蛋白相互作用;
- • 设计抗原展示纳米颗粒,用于疫苗研发;
- • 设计酶或催化 scaffold,用于新反应开发;
- • 设计小分子结合蛋白或传感器;
- • 设计多价组装体,提高结合 avidity 或免疫呈递效果;
- • 与闭环实验平台结合,实现设计-表达-测定-再设计循环。
如果说 AlphaFold 让我们更好地读取蛋白序列中的结构信息,那么 ProteinMPNN 则在很大程度上让我们能够反向书写结构所需的序列信息。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-04,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号