共折叠模型能成为药物发现底座吗?OpenDDE 给出了一种开源路线

共折叠模型能成为药物发现底座吗?OpenDDE 给出了一种开源路线

DrugIntel
发布于 2026-07-06 17:26:56
发布于 2026-07-06 17:26:56

文献来源
论文标题: Folding, Reasoning, and Scaling with Open-source Drug Discovery Engine 项目: OpenDDE Project, Aureka AI Research 研究方向: 生物分子结构预测、全原子共折叠、抗体-抗原复合物预测、AI 制药基础模型 代码链接: https://github.com/aurekaresearch/OpenDDE。
摘要
OpenDDE 试图回答一个对 AI 制药非常关键的问题:如果结构预测不只是终点,而是药物发现系统中的共享结构推理层,那么开源模型能否接近闭源前沿系统的共折叠能力?这篇工作提出一个 655M 参数的全原子生物分子基础模型,将序列、MSA、模板、原子与约束特征编码为残基级和结构 token 表示,再通过结构 token 推理、全原子扩散生成、界面形状互补损失和条件扩散训练,实现结构预测与条件设计的统一。实验上,OpenDDE 在抗体-抗原 benchmark 上取得明显优势,并展示了训练规模与测试时采样规模带来的性能提升。但它目前仍主要是一个以折叠为核心的开放结构模型,并不等同于完整 AI 药物发现系统,蛋白-小分子对接、亲和力预测、虚拟筛选和实验闭环仍有待验证。
为什么这篇论文值得关注?
AI 制药这几年有一个明显趋势:模型正在从单点任务工具,走向可以复用的生物分子基础层。
早期很多模型只解决一个窄任务,例如蛋白单体结构预测、蛋白-配体 pose 预测、抗体建模、binder 设计或亲和力预测。这样的模型有价值,但难以构成统一的药物发现平台。真正对药物发现有长期意义的模型,往往需要同时具备三种能力:理解生物分子之间的相互作用,生成全原子结构,并能够在不同条件下服务于设计、打分、优化和验证。
OpenDDE 的切入点正是共折叠。所谓共折叠,不是只预测一个蛋白单体,而是联合建模蛋白、核酸、小分子、离子、修饰残基等复杂体系的三维结构。论文把 OpenDDE 定义为一个开源、全原子、生物分子基础模型,并强调它不只是结构预测模型,而是面向后续 de novo design、亲和力估计、结构条件优化等任务的结构推理底座。
这篇文章值得关注,不是因为它声称已经完成了 AI 药物发现全流程,而是因为它把三个问题放在了一起:
第一,前沿共折叠能力能否开源化、可复现化; 第二,结构预测和条件设计能否在同一个扩散框架中统一; 第三,生物分子基础模型是否开始进入类似大语言模型的 scaling 阶段。
这三个问题都直接影响 AI 制药模型未来的工程形态。
研究背景:共折叠为什么重要?
药物发现的核心不是单个分子的孤立结构,而是分子之间的相互作用。一个抗体是否能结合抗原,一个小分子是否能进入口袋,一个 binder 是否能识别靶蛋白,一个核酸-蛋白复合物是否形成稳定界面,本质上都依赖复合物结构。
AlphaFold2 让蛋白单体结构预测达到很高水平,AlphaFold3 进一步把任务扩展到蛋白、核酸、小分子、离子和修饰残基的联合全原子建模。论文也提到,AlphaFold-style 和结构条件模型已经开始被用于表位预测、binder 设计、虚拟筛选、构象采样和实验流程解释。
但对 AI 制药而言,结构预测模型还有几个关键瓶颈。
第一,开放性瓶颈。 最强模型往往闭源,外部研究者难以独立验证训练数据、推理流程、后处理策略和 benchmark 设置。对于药物发现这种高风险场景,不能复现意味着很难建立信任。
第二,界面建模瓶颈。 抗体-抗原、蛋白-蛋白、蛋白-核酸、蛋白-小分子复合物的难点不只是把两个分子放近,而是要准确恢复界面几何:朝向是否正确,表面是否互补,原子是否冲突,局部 packing 是否合理。
第三,任务割裂瓶颈。 结构预测、条件生成、binder 设计、构象采样和亲和力估计常常被做成不同模型。这样会导致表示不统一、训练数据不能共享、下游扩展成本高。
第四,scaling 路径不清晰。 大语言模型的经验告诉我们,数据、参数和计算规模可以系统提升能力。但在生物分子结构模型中,性能提升到底来自更多数据、更大模型、更好的结构表示,还是更强推理采样,目前仍缺少足够开放的研究平台。
OpenDDE 的意义就在这里:它试图把开源共折叠、结构推理、条件扩散生成和 scaling 分析放到同一个系统中。
以往方法卡在哪里?
从技术路线看,以往方法大致可以分为四类。
单体结构预测模型:强在折叠,弱在相互作用
AlphaFold2 类型模型擅长从序列、MSA 和模板中恢复蛋白单体结构,尤其适合稳定折叠域。但药物发现更关心的是分子识别界面。单体结构正确,不等于复合物界面正确;蛋白主体 RMSD 很低,也不代表抗体 CDR loop 或配体结合口袋的局部构象足够准确。
共折叠模型:能力强,但开放性和可复现性不足
AlphaFold3 把共折叠推向全原子复杂体系。后续 Chai-1、Boltz-1、OpenFold3、Protenix 等工作也在推进开放共折叠模型。但论文认为,目前最强系统仍然闭源,训练配方、数据混合、推理策略和工程细节不透明,限制了独立验证和社区扩展。
对接与虚拟筛选模型:更接近药物任务,但结构推理层不够统一
小分子对接、亲和力预测和虚拟筛选模型直接面向药物发现,但很多方法仍把口袋结构、配体构象和打分排序拆开处理。深度学习对接模型虽然提高了 pose 采样能力,但在物理合理性、蛋白柔性、分布外泛化和筛选排序方面仍有明显不稳定性。
OpenDDE 当前并不声称解决小分子对接和虚拟筛选,它更像是提供一个上游结构底座。论文明确指出,目前模型主要聚焦蛋白-蛋白和抗体-抗原相互作用,而不是蛋白-小分子复合物;作者还观察到蛋白-蛋白界面表现与蛋白-配体 pose 预测之间相关性有限。
生成式设计模型:有设计潜力,但常与结构预测割裂
RFdiffusion、BindCraft、BoltzGen 等方法已经展示了生成式蛋白设计能力。但许多设计流程仍依赖多模型串联:一个模型生成骨架,一个模型设计序列,一个模型复核结构,一个模型打分筛选。这种流程有效,但模型之间目标不完全一致,误差会层层传递。
OpenDDE 的思路是把结构预测和条件设计统一为一个条件扩散问题:已知部分结构作为条件,未知部分作为生成目标。这个设计在概念上很重要,但当前论文对真实 de novo design 的实验验证仍然有限。
这篇论文的核心思想
OpenDDE 的核心不是简单做一个更大的 AlphaFold3 复现,而是提出一个粗到细的全原子结构生成框架。
它的主线可以概括为:
先在残基级 token 上建立全局关系,再把残基拆成更细的结构 token,经过结构推理后,用扩散模型生成全原子坐标;同时通过条件 mask,把结构预测和 de novo design 统一为同一个条件去噪任务。
这个设计有三个关键点。
第一,模型不直接从序列跳到坐标,而是在 latent space 中先推理分子关系。论文称之为 atomic latent reasoning 或 structure reasoning。残基级表示负责全局关系,结构 token 负责局部化学角色,扩散模块负责坐标生成。
第二,模型把界面几何作为专门学习目标,而不是只依赖普通坐标误差。OpenDDE 引入形状互补损失,用表面朝向、法向量关系、界面间距和 clash 惩罚来约束复合物界面。
第三,模型用 known-target atom masks 统一预测和设计。没有已知原子时,就是完整结构预测;固定一部分链、motif、口袋或结构上下文时,就是围绕条件进行目标结构生成。
5. 方法细节:OpenDDE 到底是怎么做的?
5.1 任务定义与输入输出
OpenDDE 面向的是全原子生物分子结构生成。输入可以包括:
- • 序列特征
- • 原子特征
- • MSA 特征
- • 模板特征
- • 约束特征
- • 条件生成中的 known/target atom masks
输出包括:
- • 全原子三维坐标
- • 残基级或结构级置信度
- • 距离预测相关输出
论文把 OpenDDE 描述为 coarse-to-fine molecular structure generation model。模型先编码序列、原子、MSA、模板和约束特征,形成残基 token 表示;然后扩展为结构 token;最后通过扩散坐标生成器预测全原子坐标。
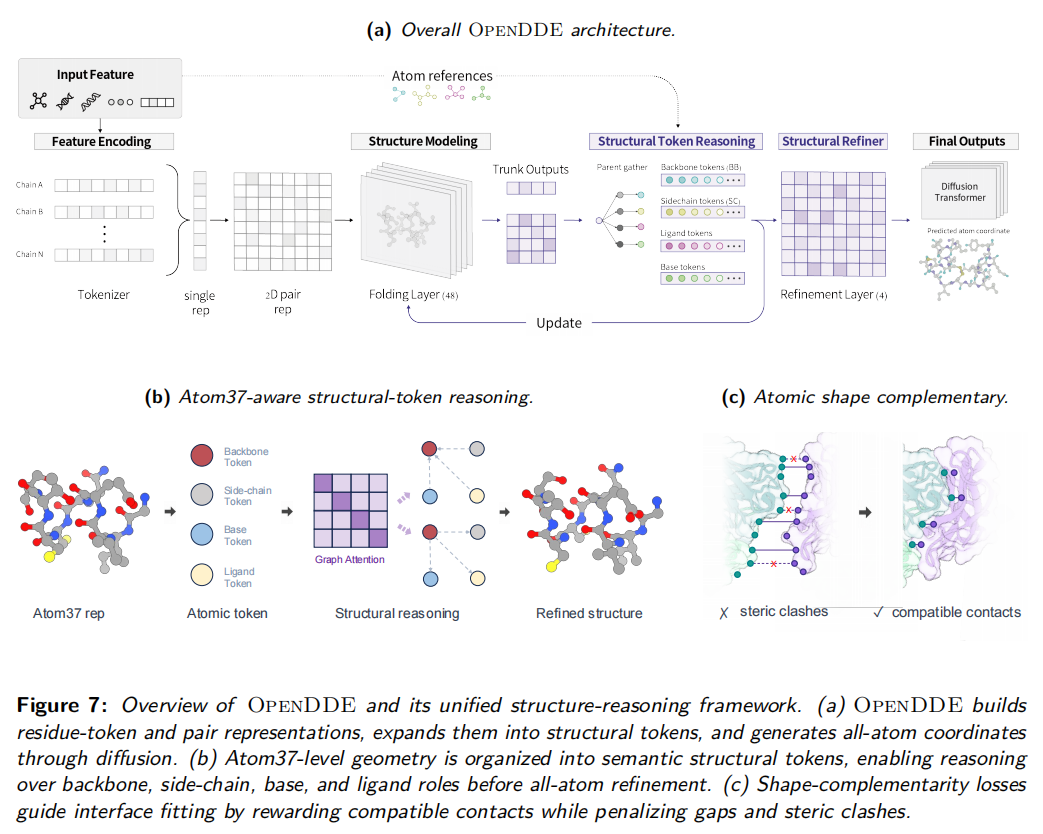
5.2 数据表示:从残基 token 到结构 token
OpenDDE 的表示分两层。
第一层是residue-token representation。这一层类似 AlphaFold 系列中的单体表示和 pair 表示:
- • 单体表示 S 记录每个 token 的上下文信息;
- • pair 表示 Z 记录 token-token 之间的关系;
- • Pairformer-style trunk 通过 single update、pair update、triangular update 等操作建模全局几何约束。
第二层是structural-token representation。这是 OpenDDE 方法中的重点。
传统残基 token 太粗,一个氨基酸被压缩成一个节点,但真实化学结构中,主链、侧链、碱基、配体原子承担的角色完全不同。OpenDDE 因此把残基或分子单元展开成更细粒度的结构 token,包括:
- • 蛋白 backbone token
- • 蛋白 side-chain token
- • 核酸 backbone token
- • 核酸 base token
- • ligand / atom token
这一步的意义在于,模型不再把一个残基看作没有内部结构的整体,而是显式区分哪些部分决定主链几何,哪些部分决定侧链 packing,哪些部分参与界面识别。
5.3 模型结构:Pairformer trunk + Structural Refiner + Diffusion Generator
OpenDDE 的主要模块如下。
第一层:Pairformer-style residue trunk
输入序列、MSA、模板、原子和约束特征后,模型先构建残基级单体状态和 pair 状态。论文报告 OpenDDE 有 655M 训练参数,Pairformer blocks 为 48,Pairformer hidden dimension 从 AlphaFold3 的 128 扩大到 384;作者指出,这会使 Pairformer 参数量约增加 3 倍,计算成本约增加 9 倍。
这个模块回答的是粗粒度问题:哪些残基可能接近,哪些链可能相互作用,哪些局部结构受到长程约束。
第二层:Structural Token Expander
模型把残基级输出扩展为结构 token 图。每个结构 token 是一个节点,两个结构 token 之间的 pair representation 是边。结构 token 初始化时不仅继承父残基表示,还加入结构角色编码。例如主链、侧链、碱基、配体 token 会拥有不同 role embedding。
论文中定义的结构 token 图可理解为:
残基级表示 → 结构角色拆分 → 结构 token 图这一层的重点是把化学角色暴露给模型,而不是让扩散模块自己从坐标损失中间接学出来。
第三层:Structural Refiner
Structural Refiner 在结构 token 图上做细粒度关系推理。它使用 pair-conditioned attention,让一个结构 token 关注其他结构 token,同时注意力权重受到 pair representation 调制。
更关键的是 triangular pair updates。三角更新的作用是让 token-token 关系不再独立更新,而是通过第三个 token 传播几何一致性。比如结构 token u 和 v 的关系,可以通过它们与 k 的关系共同修正。这对复合物界面很重要,因为真实分子结构不是两两距离的简单集合,而是大量三体、多体几何约束共同决定的结果。
第四层:Diffusion-based coordinate generator
经过结构 token 推理后,扩散模块生成全原子坐标。论文报告扩散 transformer blocks 为 24,diffusion atom encoder blocks 和 decoder blocks 各为 3。采样时 sample_diffusion.N_step 为 20,默认 sample 数为 5。
这里的扩散对象是原子三维坐标。模型不是生成 SMILES,也不是只生成残基距离图,而是在 refined structural-token features 条件下对原子坐标进行去噪。
5.4 扩散过程:预测与设计为什么能统一?
OpenDDE 的扩散训练有一个关键机制:known-target atom masks。
对每个训练样本,模型定义两类原子:
- • known atoms:作为固定结构条件输入;
- • target atoms:作为需要预测或生成的目标。
普通结构预测是特殊情况:没有 known atoms,所有 resolved atoms 都是 target。 条件设计是另一种情况:固定一部分链、motif、口袋或结构上下文,只对剩余部分加噪并生成。
训练时,真实坐标先经过随机刚体增强:
X0 → R X0 + t普通结构预测中,高斯噪声加到所有监督原子上。 条件设计中,噪声只加到 target atoms 上,known atoms 保持固定,作为结构条件参与推理。
去噪网络接收 noisy coordinates、时间步、结构 token 表示、输入特征和 known mask,预测干净坐标。扩散损失主要施加在 target atoms 上。这样,当 known mask 为空时,模型学习完整结构预测;当 known mask 不为空时,模型学习在固定上下文下生成目标区域。
这使得 OpenDDE 不需要单独设计一个 de novo design architecture。设计任务被转化为 masked molecular generation。
5.5 界面形状互补损失:让模型学会分子如何结合
只用坐标误差训练复合物模型有一个问题:两个分子可以整体距离接近,但界面朝向、局部凹凸、原子接触仍然不合理。OpenDDE 因此引入 shape-complementarity objective。
这个损失大致做了四件事。
第一,为每个 active token 定义中心点。残基 token 可以用代表性原子,侧链 token 可以用受监督原子的质心。
第二,根据同一条链周围原子密度估计局部表面法向量。直观理解,模型在判断这个 token 所处表面朝向哪里。
第三,对跨链 token pair 计算多个几何因子:
- • 两个表面是否面对彼此;
- • 法向量是否呈互补关系;
- • token 间距是否接近合理 gap;
- • 是否存在过近导致的 steric clash。
第四,把这些因子组合为 pairwise shape-complementarity score,并与真实结构中计算得到的 score 对齐。损失采用 pair-level、token-level 和 global-level 的 Huber 监督。
这一步非常关键。它让模型不是只学原子点位,而是学习界面的几何兼容性。对于抗体-抗原、蛋白-蛋白和蛋白-核酸界面,这比单纯降低坐标误差更接近真实分子识别。
5.6 全原子局部 refinement:侧链和局部坐标系不能被忽略
OpenDDE 还加入了局部全原子监督。原因很简单:全局坐标误差可以惩罚原子偏移,但不一定能很好约束残基内部几何,尤其是侧链 rotamer、核酸碱基取向和配体局部 packing。
因此模型把预测原子和真实原子变换到对应结构 token 的局部坐标系中,再计算 SmoothL1 loss。对蛋白侧链,还加入 torsion angle loss,用 1 - cos 形式约束 χ 角。对侧链和核酸碱基原子,则继承父 backbone token 的局部坐标系进行监督。
最终几何损失可以概括为:
Lgeom = Ldiff + λshape Lshape + λlocal Llocal + λsc/base Lsc/base + λχ Lχ这说明 OpenDDE 的训练目标不是单一 diffusion loss,而是把全局去噪、界面互补、局部坐标和扭转角监督组合起来。
5.7 训练数据与训练日程
OpenDDE 的训练数据遵循 AlphaFold3、Protenix 和 ESMFold2 等数据构建协议。为避免测试集泄漏,ground-truth structures 和 template databases 使用 2021-09 作为 cutoff。训练资源包括 weighted PDB、AFDB-multimer、Teddymer reproduction、MGnify long/short monomer、Swiss-Prot、disordered motifs 和 SAbDab 抗体结构。
训练采用 warmup 加四个主要阶段。整体思路是从高质量实验结构学习精确局部几何,再引入蒸馏数据扩展结构空间覆盖,最后回到高质量和任务相关数据以提升界面拟合与置信度校准。
几个细节值得注意:
- • crop tokens 从 384 增加到 544,再到 768;
- • max atoms 从 3500 增加到 6000,再到 7500;
- • SAbDab 比例在后期提高,最高达到 13%;
- • de novo design probability 在 Stage III 设为 0.1,在 Stage IV(b) 提高到 0.2;
- • Stage IV(a) 冻结主要模块,只训练 confidence head;
- • Stage IV(b) 在更大 crop size 下继续训练全模型。
计算成本也非常高:论文报告数据预处理使用 10 × 8 张 NVIDIA 80GB GPU 约五周,模型训练使用 50 × 8 张 NVIDIA 80GB Ampere GPU 六周,再用 8 × 8 张 NVIDIA 141GB Hopper GPU 训练一周,总训练成本约 414K GPU-hours。
这意味着 OpenDDE 虽然开源,但完整训练并不轻量。它更像是一个社区可复现、可扩展的基础设施,而不是普通实验室可以随意从零训练的小模型。
5.8 方法流程小结
OpenDDE 的完整流程可以概括为:
- 1. 输入序列、MSA、模板、原子特征和约束特征;
- 2. Pairformer-style trunk 构建残基级 single 和 pair 表示;
- 3. Structural Token Expander 将残基拆分为 backbone、side-chain、base、ligand/atom 等结构 token;
- 4. Structural Refiner 在结构 token 图上通过 pair-conditioned attention 和 triangular updates 做细粒度几何推理;
- 5. Diffusion coordinate generator 在 refined structural-token features 条件下去噪生成全原子坐标;
- 6. Shape-complementarity loss 约束界面朝向、间距和 anti-clash 行为;
- 7. Local-frame 和 torsion supervision 改善局部 packing;
- 8. known-target masks 将完整结构预测和条件设计统一为同一个条件扩散任务。
6. 实验设计与关键结果
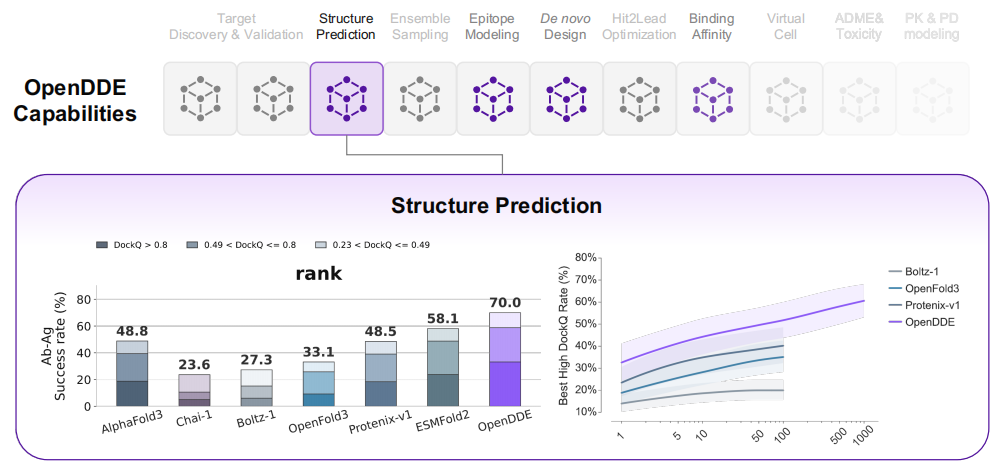
6.1 抗体-抗原 benchmark:OpenDDE 最突出的结果
论文最核心的结果集中在抗体-抗原结构预测。作者比较了 AlphaFold3、Chai-1、Boltz-1、OpenFold3、Protenix-v1、ESMFold2 和 OpenDDE,在 PXMeter-AB、FoldBench-AB 和 2026ARK-AB 三个 benchmark 上评估。
指标使用 DockQ success rate,并按照 DockQ 阈值拆分为:
- • DockQ > 0.23:acceptable;
- • 0.49 < DockQ ≤ 0.8:medium;
- • DockQ > 0.8:high quality。
在 ranking-based selection 下,OpenDDE 达到:
- • PXMeter-AB:51.0%
- • FoldBench-AB:70.0%
- • 2026ARK-AB:66.4%
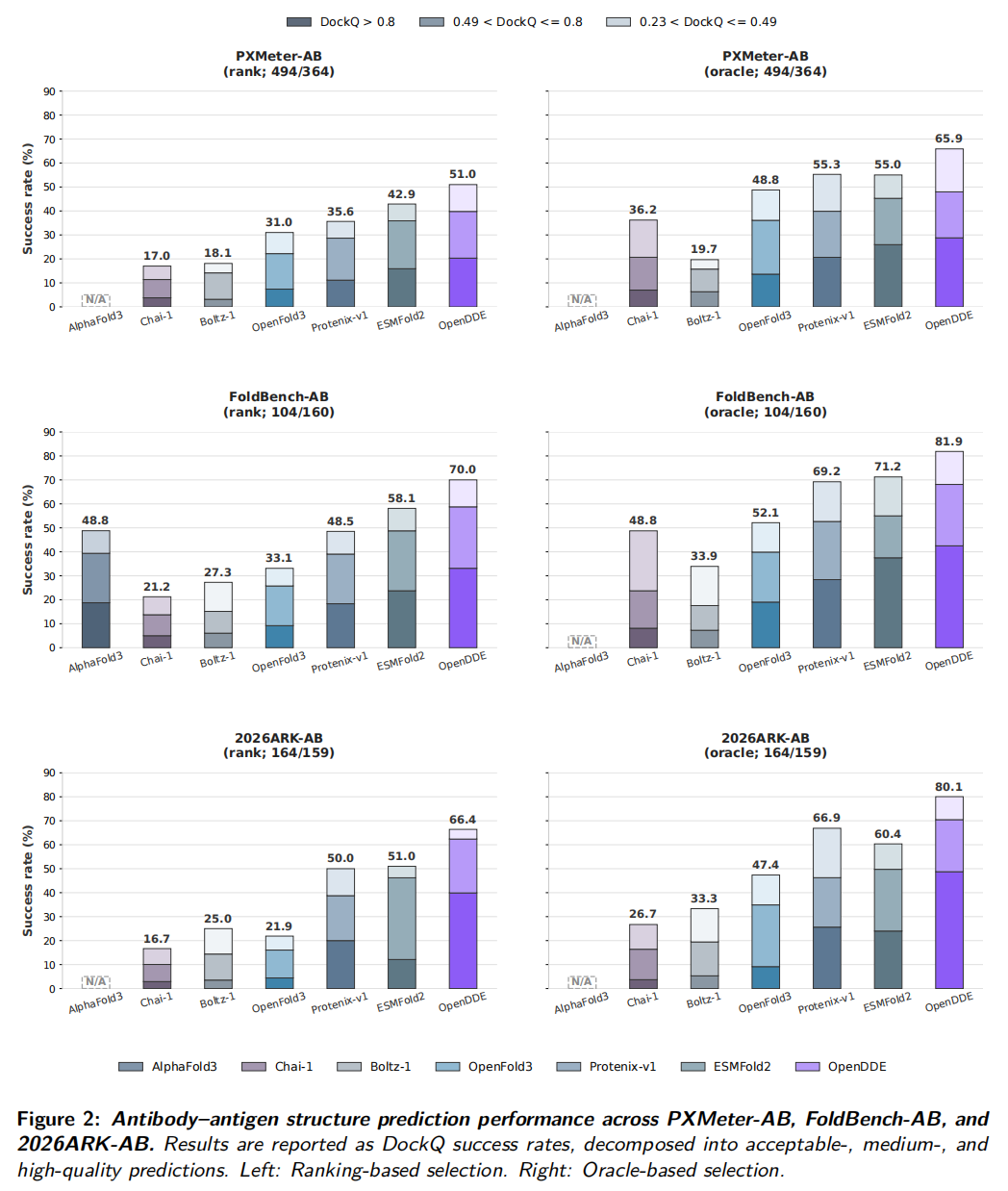
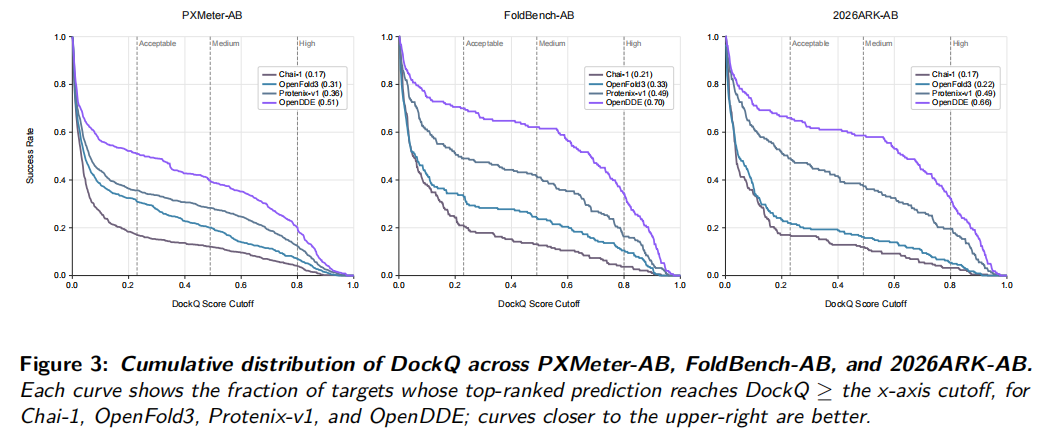
论文指出,这些提升不只是低阈值成功率提高,OpenDDE 在 medium 和 high DockQ 区间也有更高比例,说明它更好地恢复了抗体-抗原界面几何,而不是只生成勉强接近的复合物。
6.2 2026ARK-AB:强调新结构和低同源泛化
2026ARK-AB 是论文新整理的 benchmark,包含 164 个 PDB complexes,对应 159 个 unique antibody-antigen interface clusters。作者使用 SAbDab 注释抗体-抗原界面,并用 MMseqs2 easy-cluster 进行聚类,最低序列一致性阈值为 40%,alignment coverage 为 80%。这个 benchmark 旨在评估较新、低同源体系上的预测能力。
在该 benchmark 上,OpenDDE ranking success rate 为 66.4%,高于 ESMFold2 的 51.0%、Protenix-v1 的 50.0%、OpenFold3 的 21.9%、Boltz-1 的 25.0% 和 Chai-1 的 16.7%。这说明 OpenDDE 的优势在较新抗体-抗原体系上仍能保持。
6.3 Oracle 结果:模型会生成好结构,但不一定总能选出来
论文还评估 oracle selection,即从模型生成的候选中用真实 DockQ 选择最佳结构。这个设置不是实际可用性能,而是模型采样能力的上限。
OpenDDE 的 oracle success rate 为:
- • PXMeter-AB:65.9%
- • FoldBench-AB:81.9%
- • 2026ARK-AB:80.1%
这些数值明显高于 ranking-based performance,说明 OpenDDE 经常能生成高质量结构,但置信度排序或候选选择还没有完全把这些结构选出来。
这对后续开发很重要。很多模型的瓶颈不一定在采样,而在 ranking。对 AI 制药而言,生成一个正确构象但排在后面,和没有生成一样,都会影响真实使用。因此置信度校准、候选重排序和任务特异性打分可能是 OpenDDE 后续提升的关键。
6.4 Scaling laws:性能随训练规模提升,但不是只靠堆算力
论文比较了 OpenDDE、AlphaFold3、Protenix-v1、Protenix-v2、SeedFold 和 ESMFold2 在抗体-抗原预测中的表现,并用两个量估计训练规模:
- • estimated training tokens = training steps × GPUs/batch size × crop tokens;
- • estimated training cost = estimated training tokens × trainable model parameters。
OpenDDE 的 estimated training tokens 约为 2.04 × 10¹⁰,training cost 约为 1.33 × 10¹⁹。论文观察到训练规模和抗体-抗原性能之间存在正相关,并且曲线呈现平滑、次线性提升趋势。
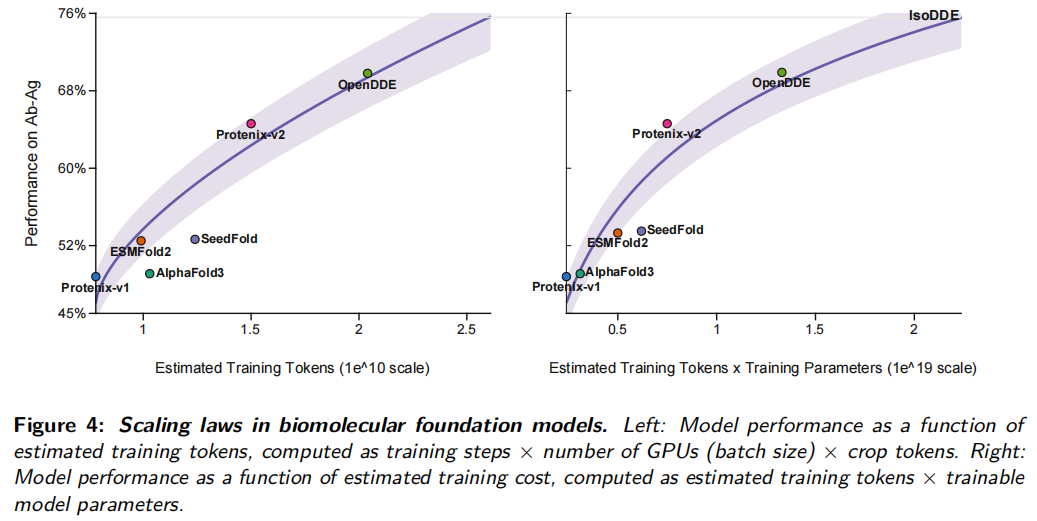
但这里不能简单理解为模型越大越好。论文也承认,不同模型偏离 scaling trend 的现象说明结构、数据质量和训练策略同样重要。对生物分子模型而言,scaling 不是单纯扩大参数,而是数据、tokenization、损失函数、推理策略和工程优化的系统扩展。
6.5 Test-time scaling:多采样确实有效,但 ranking 仍是瓶颈
OpenDDE 还测试了不同随机种子数量下的推理性能。每个 target 生成多个 stochastic samples,再用模型 ranking score 选择最终结构,同时报告 oracle 结果。
在 FoldBench-AB 上,增加 seeds 使 ranking-based high-DockQ rate 从约 28% 提升到约 34%;在 2026ARK-AB 上,从约 35% 提升到约 38%。Oracle 曲线提升更明显:FoldBench-AB 从约 66% 提升到 90% 以上,2026ARK-AB 也从约 67% 接近 90%。
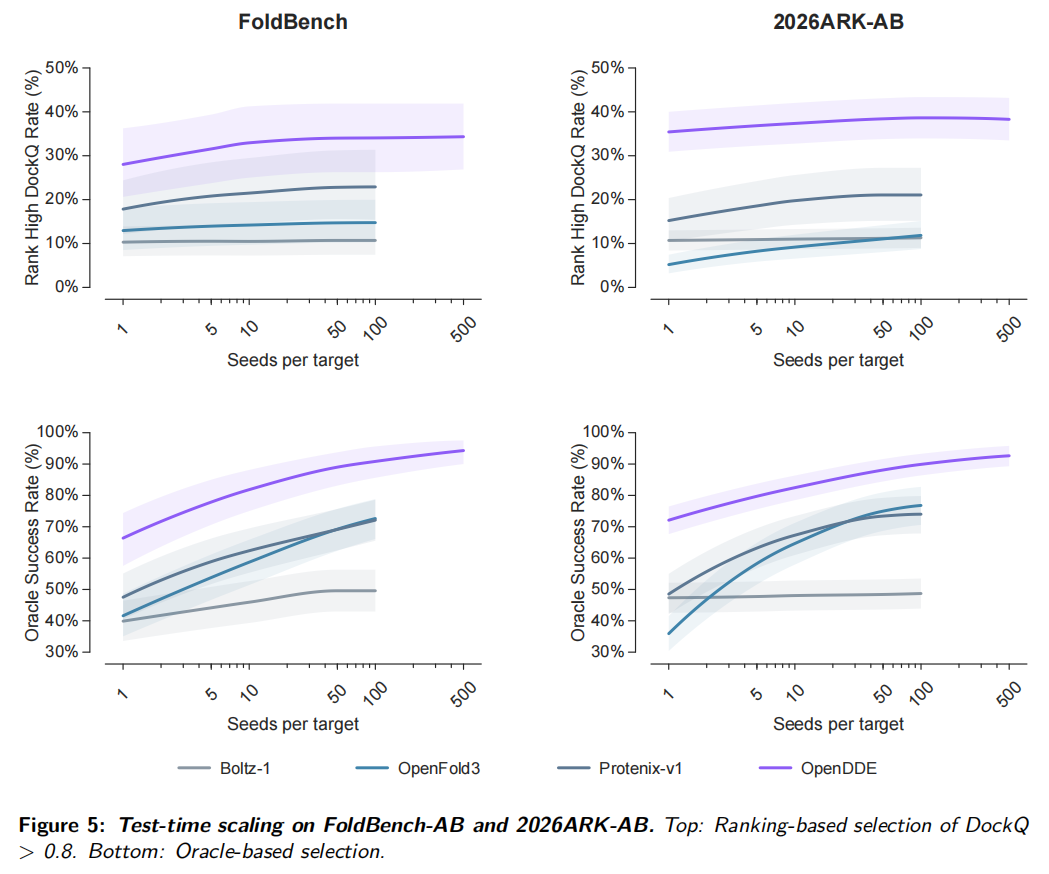
这说明测试时增加采样确实能暴露更好的抗体-抗原构象,但能否把好构象选出来仍取决于 ranking。未来如果能提高 confidence head 或重排序模型,实际性能还有明显提升空间。
6.6 FoldBench 总体任务:优势集中在 interaction-heavy systems
在 FoldBench 总体评估中,OpenDDE 在多个任务上表现强:
- • protein monomer LDDT:0.890;
- • protein-protein DockQ:0.769;
- • antigen-antibody DockQ:0.700;
- • RNA monomer LDDT:0.660;
- • protein-ligand:0.601;
- • protein-RNA:0.735。
论文认为,OpenDDE 保留了较高单链结构准确性,同时在蛋白-蛋白和抗体-抗原这类 interaction-heavy systems 上提升更明显。
这也再次说明,OpenDDE 当前最有说服力的能力不是小分子 docking,而是大分子界面共折叠,尤其是抗体-抗原结构预测。
7. 这篇文章的真正启发
启发一:共折叠模型正在从结构预测工具变成结构推理底座
OpenDDE 的定位不是单一 benchmark 模型,而是药物发现中的共享结构层。它的价值在于让不同下游任务共享同一个结构表示和扩散生成框架。
启发二:结构 token 是连接粗粒度残基表示和全原子坐标的重要中间层
直接从残基 token 到全原子坐标,中间跨度太大。OpenDDE 把残基拆成 backbone、side-chain、base、ligand/atom 等结构 token,让模型先在化学角色层面推理,再生成原子坐标。这种粗到细设计值得后续结构生成模型借鉴。
启发三:界面质量不能只靠坐标误差监督
复合物预测的关键是界面。OpenDDE 的 shape-complementarity loss 说明,模型需要显式学习表面朝向、合理间距和 anti-clash 行为。对药物发现而言,这比单纯追求 RMSD 或 LDDT 更接近真实分子识别。
启发四:采样能力和排序能力必须分开看
Oracle 结果远高于 ranking 结果,说明 OpenDDE 的生成空间里有好答案,但模型不一定能选中。这是所有生成式结构模型都需要面对的问题:生成不是终点,选择才决定真实命中率。
启发五:开源共折叠模型可能成为社区验证 scaling laws 的平台
闭源模型即使强,也难以拆解能力来源。OpenDDE 如果真正持续释放代码、权重、数据处理流程和 benchmark,会让社区更系统地研究 scaling、数据蒸馏、推理采样和结构表示之间的关系。
8. 局限性
8.1 与 IsoDDE 的比较仍然不完全透明
论文承认,由于无法获得 IsoDDE 的完整训练配方、数据混合、推理流程、后训练策略和工程优化,OpenDDE 与 IsoDDE 的比较只能在公开信息基础上进行。也就是说,即使 OpenDDE 达到类似准确率,也很难判断差异到底来自架构、数据、训练规模、蒸馏策略还是推理工程。
8.2 当前重点不是蛋白-小分子 docking
这点对 AI 制药读者尤其重要。OpenDDE 虽然名为 Drug Discovery Engine,但当前版本主要验证的是蛋白-蛋白和抗体-抗原相互作用。作者明确表示,不宣称当前模型已优化于 ligand docking、virtual screening 或 affinity ranking。
因此,不能把 OpenDDE 在抗体-抗原上的成功直接外推到小分子药物筛选。蛋白-小分子结合更依赖局部化学互补、配体构象、诱导契合、水分子、质子化状态、金属配位和打分函数,这些问题不一定能由大分子共折叠能力自动解决。
8.3 De novo design 目前更像框架能力,不是完整验证能力
论文提出结构预测和 de novo design 的统一条件扩散框架,但当前主要结果仍是结构预测,尤其是抗体-抗原共折叠。条件设计是否能产生可表达、可折叠、可结合、可实验验证的 binder 或分子组件,还需要更直接的设计 benchmark 和湿实验数据。
8.4 计算成本非常高
414K GPU-hours 的训练成本说明,OpenDDE 的完整复现门槛很高。开源代码和权重可以降低使用门槛,但社区想从头验证数据规模、训练策略和 scaling laws,仍然需要大量计算资源。
8.5 Ranking 与 confidence calibration 仍是明显短板
Oracle 与 ranking 之间的差距说明,模型生成能力没有完全转化为实际可用性能。真实应用中没有 ground truth DockQ,研究者只能依赖模型置信度、物理后处理、外部打分或实验验证。因此,如何选出正确结构,可能比继续生成更多结构更重要。
8.6 缺少面向药物发现闭环的验证
一个真正成熟的 AI drug discovery engine 至少需要连接结构预测、分子设计、亲和力评估、可合成性、ADMET、实验反馈和多轮优化。OpenDDE 当前建立的是 folding-centered foundation,不是完整闭环系统。论文也承认,分子设计、亲和力预测和实验反馈仍属于未来扩展。
对 AI 制药未来发展的意义
OpenDDE 对 AI 制药的意义,可以从三个层面理解。
第一,它推动了开放共折叠基础模型 的建设。药物发现模型不能长期依赖不可复现的黑箱系统。开源模型虽然未必立即超越闭源模型,但它能让社区验证数据、训练、推理和 benchmark,形成可信技术生态。
第二,它强调结构预测与结构条件设计的统一。未来 AI 制药模型可能不再把 prediction 和 generation 分开,而是把所有任务写成条件生成问题:给定靶点、口袋、motif、抗原、约束或候选结构,生成需要补全、优化或设计的分子部分。
第三,它让测试时计算 成为结构模型的重要变量。过去我们常把模型性能看成固定数字,但 OpenDDE 显示,多采样可以持续提高 oracle 上限。未来模型竞争可能不只是训练规模竞争,也会是 inference-time search、confidence ranking 和后处理策略的竞争。
不过,AI 制药最终不是结构预测比赛。真正转化仍要回答:预测结构能否提高命中率,生成 binder 是否可表达,设计小分子是否可合成,亲和力排序是否能指导实验,模型是否能在真实项目中缩短周期、降低成本、提高成功率。OpenDDE 为这些任务提供了一个开放结构底座,但底座之上的药物发现系统仍需要大量工程和实验验证。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号