分子生成的新接口:Sesame 用空间密度图统一口袋、片段与扩散生成

分子生成的新接口:Sesame 用空间密度图统一口袋、片段与扩散生成

DrugIntel
发布于 2026-07-06 17:28:03
发布于 2026-07-06 17:28:03

文献来源
论文标题: Sesame: Structure-Aware Molecular Generation via Spatial Density-Map Conditioning 作者: Konstantin Yatsenko, Arvind Thiagarajan 机构: Tessel Biosciences, Inc. 版本: arXiv:2606.23856v2,2026 年 6 月 24 日
摘要
这篇论文值得关注的地方,不在于又提出了一个分子生成模型,而在于它尝试解决 AI 分子生成与真实药化流程之间的一个关键错位:真实项目中更常见的任务不是从零生成一个分子,而是在已知 hit、scaffold 或 fragment 的基础上做先导优化。Sesame 的核心设计是把蛋白口袋和药化片段都表示为连续空间密度图,再通过同一个条件机制引导扩散模型同时生成原子类型、键类型和三维坐标。这样,模型既能做 de novo 生成,也能围绕药化专家保留的片段继续生长分子。实验显示,片段条件生成中 94.8% 的分子保留了种子片段,经过简单后处理后,蛋白+片段与蛋白-only 两种场景的分子有效率分别达到 92.4% 和 88.7%。但这项工作仍主要停留在生成有效性和性质分布验证阶段,距离真实活性、可合成性和前瞻性实验验证仍有距离。
为什么这篇论文值得关注?
AI 分子生成领域有一个长期矛盾:模型越来越擅长在三维空间里生成分子,但真实药物发现中的问题往往不是让模型凭空造一个漂亮分子,而是让它在已有信息的约束下做合理修改。
药化项目里,研究者通常已经有一个 hit、一个核心 scaffold、几个关键相互作用,或者一个希望保留的片段。真正困难的是:哪些位置可以长取代基?哪些相互作用应该增强?如何在不破坏核心结构的情况下改善口袋匹配、理化性质和可合成性?
很多生成模型把问题设定为 de novo ligand generation,也就是给定蛋白口袋后从零生成小分子。这个设定适合方法学展示,但和先导优化仍有距离。另一方面,scaffold decoration、fragment growing、linker design 等方法虽然更贴近药化流程,但往往把片段当作硬约束,即固定某些原子和键,再让模型补全剩余部分。这样做的问题是,模型容易被离散原子级约束卡住,难以把片段、口袋环境和整体分子几何放在一个统一的生成过程中处理。
Sesame 的切入点正是在这里:它把蛋白口袋和片段都转换为空间密度图,让二者成为同一种连续条件信号。模型不再分别设计一套 pocket-conditioning 模块和 fragment-conditioning 模块,而是用一个统一的密度图接口让生成模型学会:哪里有疏水区域,哪里有氢键供受体,哪里存在范德华排斥或空间占位,哪里是药化专家希望保留的片段。
研究背景
基于结构药物设计中的生成模型大致面临三类瓶颈。
第一类是表示瓶颈。蛋白口袋是三维物理环境,包含形状、静电、疏水性、氢键、芳香相互作用和空间排斥等信息。传统口袋条件生成方法通常把蛋白原子作为图或点云输入,这能保留原子级结构,但模型需要自己从原子类型和坐标中学习复杂的局部物理场。Sesame 选择把口袋预先转成多通道密度图,相当于把一些物理化学信息显式放入空间网格中。
第二类是任务瓶颈。de novo 生成强调从零探索化学空间,但先导优化更强调在已有结构基础上的局部修改。很多模型可以生成新分子,却不一定能尊重药化专家指定的核心片段。Sesame 明确把 fragment-conditioned generation 当作核心任务,而不是附属功能。
第三类是扩散对象瓶颈。小分子既有连续三维坐标,又有离散原子类型和离散键类型。只扩散坐标会丢掉化学图信息,只扩散分子图又难以生成合理三维构象。Sesame 因此设计了一个混合扩散过程:坐标用高斯扩散,原子类型和键类型用 categorical diffusion,并在反向过程中联合去噪。
以往方法
已有方法可以分成几条路线。
第一条路线是三维点集扩散。 这类模型直接在原子坐标和原子类型上做扩散,强调 E(3)/SE(3) 等变性,适合生成三维分子构象。但许多方法需要在采样前固定原子数,难以自然表达长出几个原子、删掉几个原子、从片段继续扩展这类药化操作。
第二条路线是口袋条件生成。 TargetDiff、DiffSBDD 等方法将蛋白结合位点作为条件,直接在口袋中生成配体。它们推动了 structure-based de novo design,但通常更关注从口袋到新配体,而不是从已有 hit 或 scaffold 出发做先导优化。
第三条路线是 scaffold decoration、fragment linking 和 fragment growing。 这类方法更贴近药化,但很多做法把 scaffold 或 fragment 固定为离散原子结构,模型围绕硬约束做补全。硬约束很清晰,但不够柔性:真实药化中保留的可能是空间形状、关键相互作用或局部药效团,而不总是完全固定每个原子的三维位置。
第四条路线是空间场或 voxel 表示。 这类方法把分子或口袋表示成三维空间场,例如形状、电势、药效团或相互作用密度。Sesame 吸收了这条路线的思想,但它的关键不是单纯使用 voxel,而是用同一种密度图条件同时表达口袋和片段,并通过 Pairformer 式结构让点集分子生成模型主动读取这些空间场信息。
Sesame 的核心思想:把口袋和片段统一成空间条件
Sesame 的中心假设可以概括为一句话:
如果蛋白口袋和药化片段都能被表示为连续空间密度场,那么模型就可以用同一套条件机制完成 de novo 生成和片段引导的先导优化。
这个设计有两个重要含义。
第一,片段不再只是固定原子集合,而是一个空间先验。药化专家可以把一个 hit 剪成想保留的核心结构,Sesame 将这个片段转成 ligand density map,再和 protein density map 一起输入模型。模型看到的不是必须一动不动的离散 scaffold,而是一个连续场:这里已有结构占位、这里有相互作用特征、这里应该围绕它继续生长。
第二,蛋白口袋和片段共享同一种条件语言。蛋白口袋提供外部环境,片段提供内部起点,二者都进入密度图条件模块。模型不需要为 pocket 和 fragment 写两套独立结构,而是在同一个空间读取机制中学习如何利用这两类信息。论文强调,这使得 Sesame 同时支持 protein-only de novo design 和 protein+fragment lead optimization。

5. 方法细节:Sesame 到底是怎么做的?
5.1 任务定义与输入输出
Sesame 的生成对象是一个最多包含 50 个重原子的三维小分子。模型输入包括:
- • 当前扩散步下的 noisy atom types;
- • noisy atomic coordinates;
- • noisy bond types;
- • 三个独立噪声水平:坐标噪声、原子类型噪声、键类型噪声;
- • 可选的 protein density map;
- • 可选的 ligand 或 fragment density map;
- • 用于控制条件是否存在的 density gates。
模型输出三个结果:
- • 每个潜在原子位置的原子类型 logits;
- • 每个原子的三维坐标;
- • 每对原子之间的键类型 logits,并通过对称化保证 i-j 和 j-i 的键一致。
为了支持可变原子数,Sesame 不直接让模型决定生成多少个原子,而是设定最大原子数 N=50,并增加一个 pseudo-None atom type。最终不存在的原子位点由模型预测为 None。这是一个实用但不完美的设计:它让固定维度网络可以生成不同大小的分子,但也带来了论文中观察到的 spurious atoms 问题。
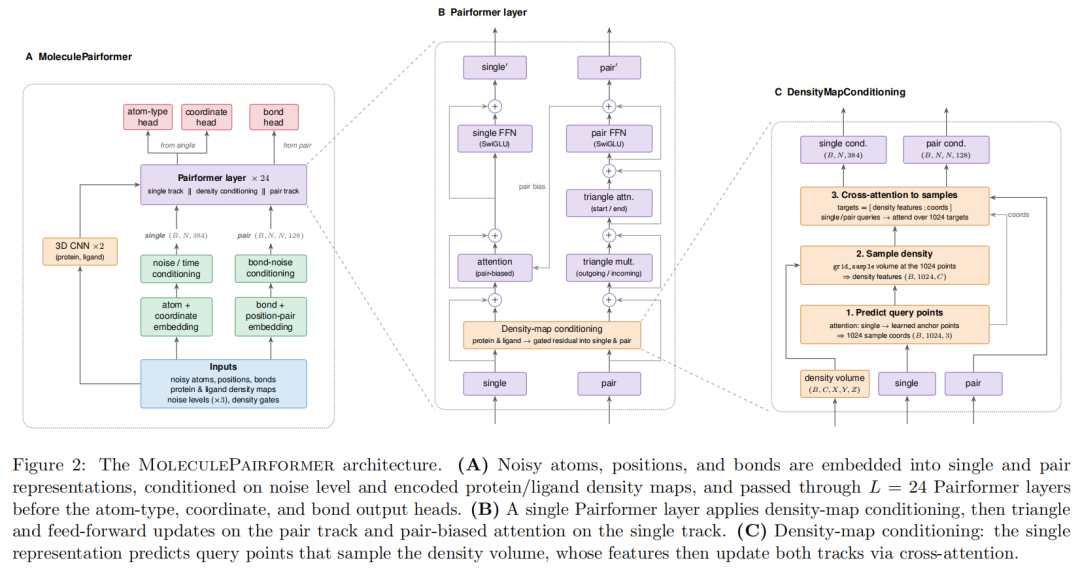
5.2 数据表示:原子点集 + 原子对表示 + 空间密度图
Sesame 的主体网络叫 MoleculePairformer,包含两类核心表示。
第一类是 single representation,即每个原子的表示,维度为 N × 384。它承载单个原子槽位的信息,包括当前 noisy atom type、三维位置、扩散时间等。
第二类是 pair representation,即每对原子的表示,维度为 N × N × 128。它承载原子对之间的相对几何和键类型信息。这个设计明显借鉴了 AlphaFold 系列中的 Pairformer/Evoformer 思路:如果分子不是孤立原子的集合,而是原子对关系高度约束的结构,那么 pair track 对建模键、距离、角度和整体拓扑非常重要。
原子类型不是简单的元素标签,而是元素与隐式氢数的组合。论文使用 16 类常见类药重原子状态,例如 C0、C1、C2、C3、N0、N1、N2、O0、O1、F、P、S、Cl、Br、I 等,再加上 pseudo-None,总计 17 类。这样做的好处是,模型不显式生成氢原子,也能通过隐式氢数编码局部价态信息。
键类型包括 single、double、triple、aromatic、None 等。因为绝大多数原子对之间没有键,所以 bond diffusion 中 None 类占据极大先验权重。
密度图是 Sesame 的关键条件输入。每个 protein 或 ligand density map 是 6 × 32 × 32 × 32 的三维网格,分辨率 0.5 Å,立方体边长 16 Å。6 个通道分别编码 charge、hydrophobicity、H-bond donor、H-bond acceptor、aromatic interactions 和 van der Waals potential。之后密度图经过 3D CNN 和 MaxPool,变为 384 × 12 × 12 × 12 的体素特征。
5.3 密度图条件模块:模型主动决定看哪里
Sesame 最有意思的模块是 DensityMapConditioning。
普通 voxel 模型往往把整个网格直接送进 3D CNN,然后让模型全局消化。但 Sesame 的分子主体仍是原子点集和原子对表示,因此它需要一个机制把体素场信息转移到原子表示中。
具体做法分三步。
第一步,模型从 single representation 中预测一组采样点。每个 Pairformer layer 都会产生采样坐标。论文设定 4 个 attention heads,每个 head 产生 256 个采样点,总共 1024 个采样点。可以理解为:当前分子雏形根据自身状态,主动在口袋或片段密度场中选择需要读取的位置。
第二步,用 grid_sample 对密度图做三线性插值,提取这些采样点处的 density features。这样模型不需要遍历整个 3D 网格,而是用可学习方式读取与当前生成状态最相关的空间区域。
第三步,把采样点坐标和密度特征拼接起来,作为 cross-attention 的 key/value,分别更新 single representation 和 pair representation。更新结果通过 gated residual connection 加回原始表示。
这个设计的价值在于,它把体素条件从静态背景变成了可查询的空间记忆。模型不是被动接收口袋,而是在每一层根据当前原子和原子对状态,决定从密度场中提取什么信息。
5.4 MoleculePairformer:用单体轨道和成对轨道共同建模分子
每个 Pairformer layer 包括几类操作:
- • density-map conditioning:从口袋/片段密度图读取空间信息;
- • triangle multiplication 和 triangle attention:更新原子对表示,捕捉 i-j-k 三体关系;
- • pair-biased attention:用 pair representation 作为 attention bias 更新 single representation;
- • SwiGLU feed-forward network;
- • residual connection 和 pre-layer normalization。
这套结构的逻辑很清楚:分子生成不是只预测每个原子在哪里,而是要同时满足局部价态、键连关系、三维距离、整体拓扑和口袋几何约束。single track 负责原子级信息,pair track 负责原子对关系,density conditioning 负责把外部空间场注入两条轨道。论文使用 24 层 Pairformer,single 维度 384,pair 维度 128,attention heads 为 4。
5.5 混合离散-连续扩散:坐标、原子类型、键类型一起去噪
Sesame 的扩散过程不是单一变量扩散,而是三个变量共同扩散:
- • 连续坐标 C;
- • 离散原子类型 A;
- • 离散键类型 B。
坐标使用 Gaussian diffusion。前向过程向真实坐标加高斯噪声,噪声强度由 power-law schedule 控制,最小噪声 0.01 Å,最大噪声 8 Å。一个重要细节是,原子点集本质上是无序的。加噪后,第 i 个 noisy point 未必还对应真实分子中的第 i 个原子。为了解决这个对应关系问题,作者使用 modified Jonker-Volgenant assignment,把 noisy points 与 ground-truth atoms 重新匹配,最小化总位移。这个匹配不移动点,只重新定义哪个 noisy point 应该向哪个真实原子去噪。随后,原子类型和键矩阵也按同一置换重新排列。
原子类型使用 categorical marginal diffusion。噪声水平由余弦 schedule 控制。当噪声为 0 时,保持真实类型;当完全加噪时,原子类型趋向均匀分布。这样做避免模型在 full noise 状态下天然偏向训练集中更常见的原子类型。
键类型也使用 categorical diffusion,但先验不是均匀的。因为分子图中大多数原子对没有键,作者给 None bond 设定 0.99 的先验权重,而 single/double/triple/aromatic 各给很小但非零的权重。这一设计符合分子图稀疏性,也避免反向采样时合法键类型概率变成严格为零。
反向过程中,网络预测 clean atom type logits、clean bond logits 和 clean coordinates。坐标使用 DDIM 风格的 deterministic update;原子类型和键类型根据模型预测的 x0 分布以及离散扩散的贝叶斯后验进行采样。论文特别强调,在离散反向后验中,一个与 q(aτ|a0) 相关的分母不能从求和中拿出来,否则会在低噪声阶段把概率过度集中到主类,伤害少数类别预测。这个细节看似数学化,但对化学生成很重要,因为少数原子类型或少数键类型经常决定分子的真实化学多样性。
5.6 训练目标:不只预测类型,还约束几何关系
训练时,Sesame 从真实分子出发随机采样三个噪声水平,得到 noisy atom types、noisy bonds 和 noisy coordinates,然后让模型预测去噪后的原子类型、键类型和坐标。
损失函数包括四部分:
- • atom type cross-entropy,带 0.02 label smoothing;
- • bond type cross-entropy,带 0.001 label smoothing;
- • coordinate MSE;
- • LDDT-like loss,用 0.5、1.0、2.0、4.0 Å 四个阈值度量原子对距离是否接近真实结构。
总损失是四项简单相加,没有额外权重。LDDT-like loss 的意义在于,它不只要求单个原子坐标接近,还要求多原子距离关系合理,从而间接约束键长、键角、二面角和整体几何。
5.7 数据与增强:从大规模可购买分子到合成结构复合物
Sesame 使用两类数据。
第一类是 ZINC22 ligand-only 数据。作者称 ZINC22 包含超过 370 亿个 ready-to-make 商业可获得化合物,他们下载了约 150 亿个用于训练,并筛选 3–50 个重原子的分子。每个分子用 RDKit 生成 3D conformer,经过 MMFF94 初始化,再计算 ligand density map。
第二类是 SAIR protein-ligand 数据,约 800 万个蛋白-配体复合物,论文说明其主要是 co-folded synthetic structures,而不是实验复合物。处理时,作者以配体周围一定半径提取结合口袋,计算蛋白原子的 charge、hydrophobicity、H-bond donor/acceptor、aromaticity、VdW radii,并生成 protein density map。
数据增强包括两类。
一类是随机三维旋转,用 QR decomposition 生成 SO(3) 上的随机旋转,避免模型记住固定坐标方向。
另一类是 fragment augmentation。作者用 RDKit 的 Murcko scaffold 作为起点,随机采样目标片段大小。如果目标片段小于 scaffold,就随机删原子,但不能让片段断裂;如果目标片段大于 scaffold,就从原分子中逐个加回与当前片段相连的原子。这样得到一组大小分布较均匀、但偏向 scaffold-like 的片段。在 50% ligand-conditioned cases 中,这个片段用于生成 ligand density map。
5.8 轨迹微调:让训练更接近真实采样
扩散模型训练通常是在随机噪声水平下做单步去噪,但真实生成是多步反向采样。两者之间存在 train-sampling mismatch。Sesame 用 trajectory finetuning 处理这个问题。
具体流程是:先按预训练方式采样数据点,把它加噪到随机噪声水平;然后从这个状态开始运行 100 步反向扩散,记录每个中间状态;再把最终预测与 ground truth 做匹配,用匹配得到的置换重排真实分子;最后把每个中间状态和重排后的真实目标组成训练样本继续微调。
这个设计很关键,因为模型在采样早期可能会交换原子槽位。如果仍用原始 ground truth 顺序监督,会让训练目标和模型轨迹错位。重新匹配相当于承认生成轨迹中的原子身份会发生置换,并让监督信号跟上这种置换。
5.9 实际使用流程
如果用于先导优化,一个实际流程可以是:
- 1. 药化专家从已有 hit 中选择希望保留的 scaffold 或 fragment;
- 2. 将蛋白口袋转成 protein density map;
- 3. 将 fragment 转成 ligand density map;
- 4. Sesame 从高噪声状态开始联合生成原子类型、键类型和三维坐标;
- 5. 对生成分子做后处理,去除孤立 spurious atoms;
- 6. 继续进行价态检查、性质过滤、对接/重打分、可合成性评估和实验验证。
如果用于 de novo design,则可以只输入 protein density map,让模型在口袋条件下生成新分子。论文中最强调的两种药物发现场景,正是 protein+fragment lead optimization 和 protein-only de novo generation。
6. 实验设计与关键结果:结果说明了什么?
Sesame 的验证主要围绕不同条件模式展开:
- • P+L:蛋白密度图 + 完整配体密度图;
- • P+F:蛋白密度图 + 片段密度图;
- • P:仅蛋白密度图;
- • L:仅完整配体密度图;
- • F:仅片段密度图;
- • U:无密度图。
评价指标包括两类。
第一类是 single-step denoising metrics,例如 type accuracy、bond accuracy、true bond accuracy、bond precision、bond recall、position MSE 和 LDDT。这些指标衡量模型在随机部分加噪输入下,一步预测干净结构的能力。
第二类是 20-step reverse diffusion validation metrics,包括 single-fragment、atom validity 和 molecule validity。这里 single-fragment 指生成分子是否为连通单片段;atom validity 指原子价态是否合理;molecule validity 指整个分子同时满足连通性和原子有效性。论文指出,对于可变原子数模型,生成完整连通分子是一个明显挑战。
6.1 条件信息确实有用,但不同指标读法不同
在预训练模型中,P+L 条件下 single-step 指标最好,例如 type accuracy 0.983、position MSE 0.022、LDDT 0.982。这并不意外,因为完整 ligand density map 几乎提供了目标分子的强结构先验。相比之下,protein-only 条件更难,因为模型必须从口袋环境中生成完整小分子。
但从 20-step 生成轨迹看,差异没有 single-step 那么简单。预训练时,molecule validity 在 P+F、P、P+L、U 等条件下分别约为 0.776、0.773、0.755、0.734。也就是说,完整配体条件虽然有助于一步重构,但不一定直接带来最高的完整生成有效率。这提示我们:生成质量不仅取决于条件信息多少,也取决于反向轨迹是否稳定。
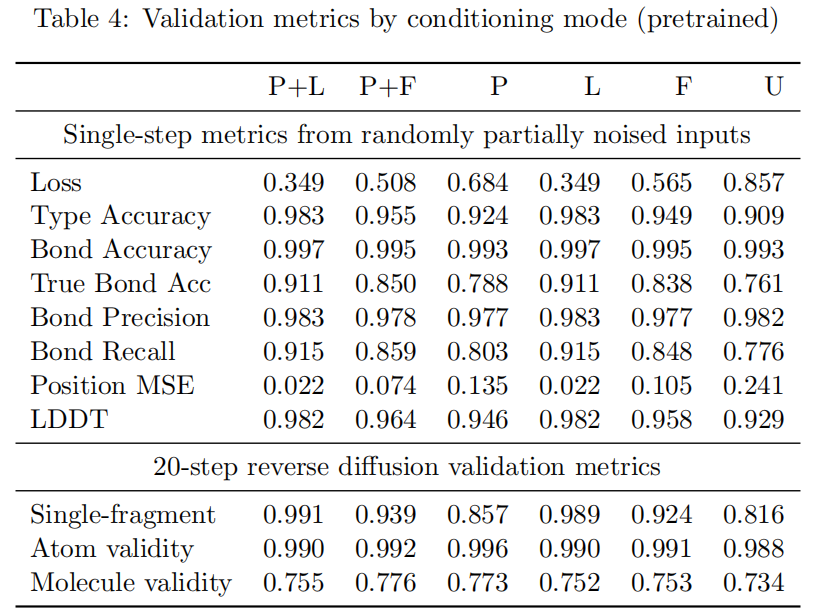
6.2 轨迹微调改善了多步生成
微调后,trajectory-level 指标整体改善。protein-only 的 molecule validity 从 0.773 提升到 0.825,P+F 从 0.776 提升到 0.805,unconditioned 从 0.734 提升到 0.763。single-fragment 指标也改善明显,例如 P+F 从 0.939 提升到 0.964,P 从 0.857 提升到 0.899。
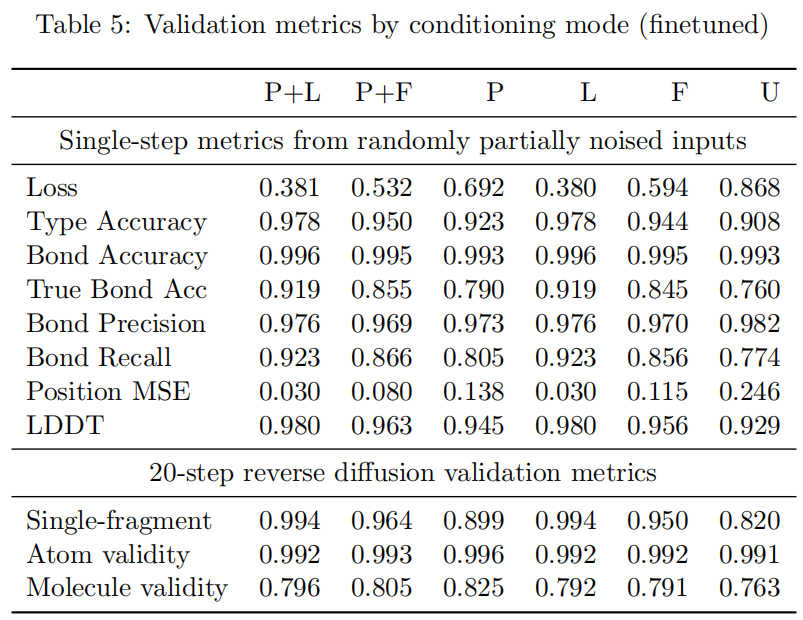
这说明 trajectory finetuning 的价值主要体现在生成轨迹层面,而不是单步去噪层面。论文也指出,微调后 single-step metrics 大致持平或略降,但 trajectory-level metrics 改善。这是合理的:微调目标更接近真实采样过程,而不是单步重构。
6.3 片段条件没有被模型忽略
对 lead optimization 来说,关键问题不是模型能不能生成有效分子,而是它是否尊重输入片段。论文报告,在所有 fragment-conditioned generation 中,包括 P+F 和 F,94.8% 的生成分子包含 seeding fragment 作为 substructure。这是 Sesame 论证片段密度图作为软先验有效的核心证据。
不过这里也要注意:保留片段不等于生成分子活性更好,也不等于修饰方向药化合理。它证明的是条件没有被忽略,而不是先导优化已经成功。
6.4 后处理大幅提高有效率,但也暴露了模型问题
论文指出,模型主要错误是生成 spurious atoms,尤其在片段条件场景中明显。这些多余原子通常是没有任何键连接的单原子。经过简单后处理去除这些原子后,P+F 和 P 两种核心药物发现场景的 molecule validity 分别达到 92.4% 和 88.7%。
这个结果有两面性。一方面,错误类型比较简单,后处理可修复;另一方面,这也说明模型内部的可变原子数控制还不够干净。pseudo-None 机制虽然让固定最大原子数模型可以生成不同大小分子,但它并没有完全解决哪些原子应该存在这一问题。
6.5 药物样性质分布接近 SAIR 配体,但这不是活性验证
论文比较了 Sesame 生成分子与 SAIR ligands 的性质分布,包括 molecular weight、cLogP、TPSA、QED、heavy-atom count、fraction Csp3 等,并用 Lipinski Rule of 5 阈值作为参考。Figure 3 显示,Sesame 生成分子的性质分布大体接近 SAIR 配体,其中 fragment-seeded generation 略优于 de novo generation。
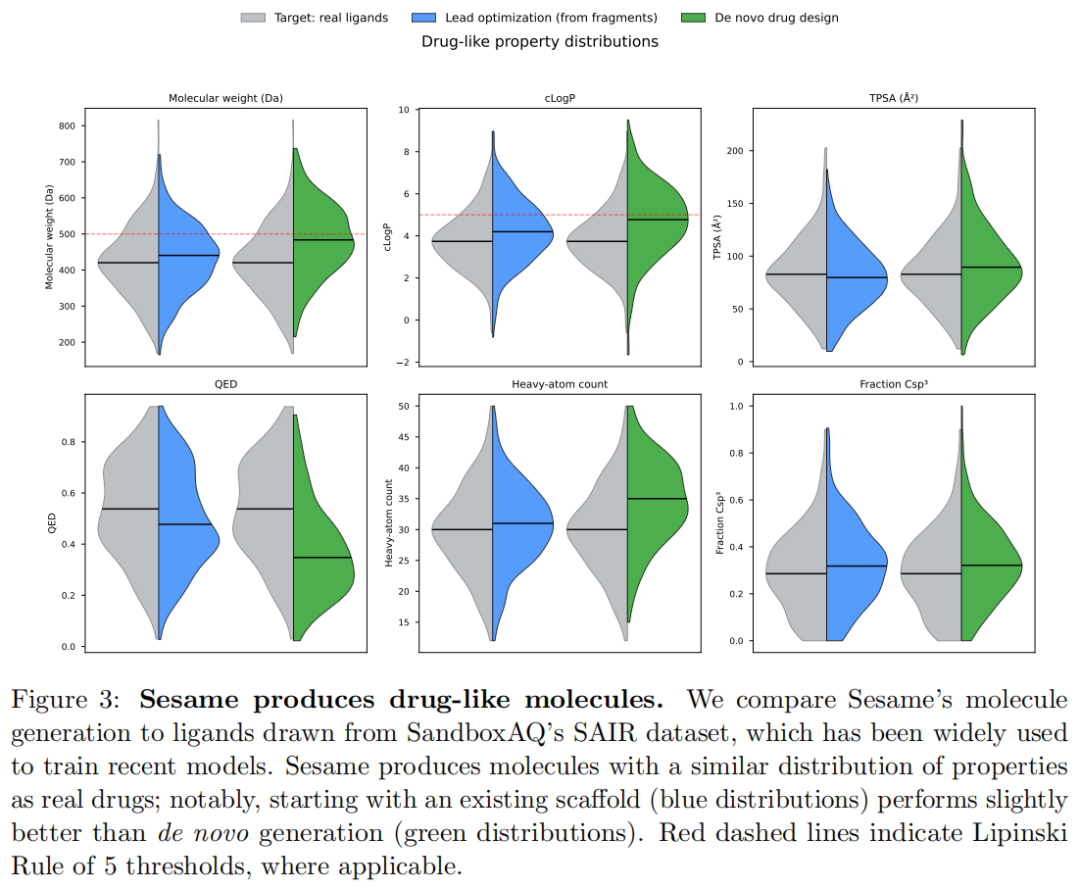
这说明 Sesame 没有生成大量明显偏离药物样分布的分子。但这类性质分布验证只能说明生成分子在统计性质上类似训练/参考集合,不能说明它们真的结合目标、具备 SAR 价值、可合成,或能在实验中显示活性。
6.6 采样路径很重要:先定几何,再定类型?
Sesame 的扩散噪声空间有三个维度:坐标、原子类型和键类型。论文对噪声 schedule 做了 sweep,用 20 步采样、64 种 gamma 组合、2048 个随机初始状态评估采样质量。结果显示,较大的 position gamma 相比 atom gamma 更优,作者解释为先把位置固定下来,再确定原子类型更合理。更有意思的是,较大的 bond gamma 也有利,意味着较早确定大部分非键关系和整体键矩阵,可以为坐标与原子类型去噪提供结构偏置。
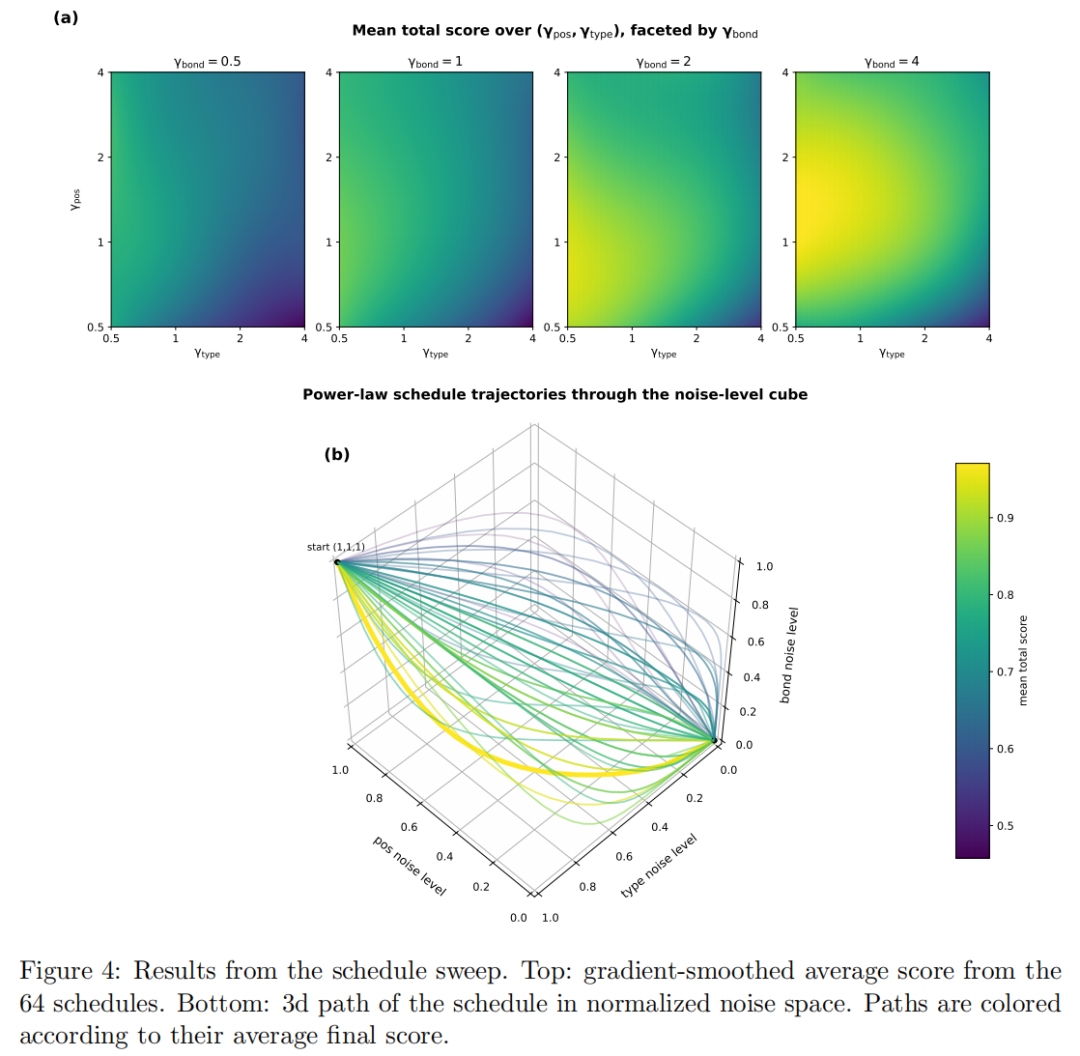
这点对后续分子扩散模型有启发:小分子生成不是所有模态同步去噪就最好。键拓扑、三维几何和原子类型之间可能存在更优的解耦节奏。
启发
第一,生成模型需要更贴近药化操作。 Sesame 的价值不是单纯生成新分子,而是把 fragment growing 放到和 de novo design 同等重要的位置。这更接近真实项目中药化专家与 AI 模型协同的流程。
第二,连续空间场可能成为结构感知生成的重要接口。 图表示适合保留离散结构,密度图适合表达空间物理环境。Sesame 试图用密度图把蛋白口袋、片段和物理相互作用统一起来,这比单纯把蛋白原子坐标喂给模型更直接。
第三,混合扩散不能只看模型架构,也要看反向采样数学。 论文对离散反向后验的细节、原子匹配、bond prior 和 noise schedule 都做了专门处理。这提醒我们,在小分子生成中,很多质量问题不一定来自网络不够大,而可能来自扩散变量定义和采样路径不合理。
第四,trajectory finetuning 是值得关注的方向。 真实采样轨迹和训练时随机加噪状态不同。让模型在自己的 rollout 上继续学习,有助于减少多步采样累积误差。
局限性
这篇工作方法设计有新意,但证据边界也比较清楚。
第一,缺少真实药物发现闭环。 论文主要验证分子有效性、片段保留率和性质分布,没有报告 docking enrichment、binding affinity correlation、合成路线、体外活性或前瞻性实验。因此它证明了模型可以生成看起来合理且尊重片段的分子,但还没有证明这些分子能成为真实 hit 或 lead。
第二,对比基线不足。 论文相关工作讨论了 TargetDiff、DiffSBDD、FLOWR、ShEPhERD、DiffLinker、DiffDec 等方法,但实验部分主要是 Sesame 内部不同条件模式的验证,缺少与强基线在同一 benchmark 上的系统比较。因此很难判断 Sesame 相比现有口袋条件生成或 fragment-based generation 的实际优势有多大。
第三,SAIR 数据的性质需要谨慎看待。 SAIR 被描述为主要由 co-folded synthetic structures 构成,而不是大规模实验解析复合物。用这类数据训练结构生成模型可以带来规模优势,但也可能引入 co-folding 模型本身的偏差。生成分子性质接近 SAIR ligands,并不等价于接近真实药物项目中的活性化合物分布。
第四,密度图表示有压缩损失。 空间密度图可以表达局部物理环境,但会弱化残基身份、蛋白序列上下文、局部构象异质性等因素。论文中的 protein density map 对蛋白重原子使用 hardcoded parameters,且不根据局部环境调整,这对复杂结合位点可能不够。
第五,化学有效性仍依赖后处理。 spurious atoms 是论文明确提到的主要错误。虽然可以通过去除孤立原子改善有效率,但理想生成模型应尽量从采样过程中减少这类错误,而不是依赖后处理修补。
第六,合成可及性尚未内生化。 论文也承认,生成结构有效分子不等于可合成。Sesame 目前是 atom-level generation,并未直接在 building block、reaction template 或 retrosynthesis-constrained space 中生成。后续如果用于真实项目,需要接入合成可及性过滤或反应约束生成。
对 AI 制药未来发展的意义
Sesame 的方向有三个潜在影响。
第一,它推动结构感知分子生成从纯 de novo 走向 human-in-the-loop lead optimization。未来更有价值的 AI 工具可能不是替代药化专家,而是让药化专家指定保留结构、关键相互作用或空间约束,模型负责快速生成可比较的局部改造方案。
第二,它提示 pocket、fragment、pharmacophore、interaction field 可以被统一成空间条件接口。如果这一思路继续发展,未来模型可能输入多种空间场:蛋白口袋场、已知配体场、药效团场、排斥场、亲和力热点场、合成可达片段场,再由生成模型综合这些条件。
第三,它为分子扩散模型提出了更细的设计问题:不同模态应该如何加噪和去噪?坐标、键、类型、片段约束、口袋条件之间应该同步更新,还是按某种节奏逐步确定?Sesame 的 schedule sweep 说明,这类问题不是实现细节,而会直接影响生成质量。
但真正走向 AI 制药应用,还需要把 Sesame 这样的生成引擎放入更完整的闭环:生成、过滤、对接/重打分、MD 或物理校正、合成路线评估、实验测试、活性反馈和再训练。只有进入这个闭环,模型生成能力才可能转化为发现能力。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-05,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号