Nat. Commun. | 双图表示增强Transformer模型的分子性质预测

Nat. Commun. | 双图表示增强Transformer模型的分子性质预测

DrugAI
发布于 2026-07-13 16:13:15
发布于 2026-07-13 16:13:15
DRUGONE
准确预测分子性质是化学、材料科学和药物研发中的核心问题,其性能高度依赖于分子表示方式。近年来,Transformer模型凭借全局自注意力机制逐渐应用于分子学习,但如何充分编码原子、化学键、拓扑结构及立体几何信息,仍然是限制模型性能的重要因素。研究人员提出了一种名为Dual Graph Transformer(DGT)的新型深度学习框架,通过同时构建原子图和键图两种互补表示,在自注意力机制中联合建模原子特征、键特征、图拓扑、环结构以及三维立体信息,实现更加完整的分子表示。
研究人员在MoleculeNet十个数据集、58项分子性质预测任务上进行了系统评估。结果表明,DGT在药物毒性、生物活性、理化性质和量子化学性质预测方面均优于现有主流模型。同时,该模型不仅能够融合三维几何信息和立体化学信息进一步提升预测精度,还能够通过注意力机制识别影响分子性质的重要结构片段,为模型提供良好的可解释性。

分子性质预测广泛应用于药物发现、催化剂设计、新材料开发等领域。然而,传统实验测量和第一性原理计算成本较高,难以满足海量化学空间筛选需求。随着深度学习的发展,越来越多的模型被用于自动学习分子结构与性质之间的关系。
目前常见的分子表示包括分子指纹、SMILES字符串以及分子图。其中,图神经网络能够直接利用原子和化学键构建分子图,因此成为分子机器学习的重要方向。然而,传统图神经网络依赖局部消息传递机制,随着网络加深容易出现过平滑问题,且只能学习有限邻域的信息,难以充分建模复杂分子的长程依赖关系。
Transformer的自注意力机制突破了这一限制,可以直接建立任意两个节点之间的联系,因此逐渐成为分子表示学习的重要发展方向。然而,分子并非自然语言,其结构具有复杂拓扑和空间几何特征,仅利用原子图仍无法充分表达化学键之间的相互作用。因此,如何设计更加符合化学规律的分子表示,是进一步提升Transformer性能的关键问题。
方法
研究人员提出了Dual Graph Transformer框架,将同一分子同时表示为原子图和键图。原子图以原子作为节点、化学键作为边;键图则通过线图变换,将化学键视为节点,原子连接关系转化为边,从而从另一种角度描述分子拓扑结构。
在模型中,原子图和键图分别建立独立的自注意力模块,并通过特征融合实现信息交互。模型同时引入最短路径距离编码、随机游走位置编码以及环结构编码,使Transformer能够感知分子拓扑关系。此外,模型还支持三维几何信息,包括键长、原子间距离、键角,以及R/S手性和E/Z异构信息,从而实现二维拓扑与三维结构的统一建模。最终,经过多层图Transformer编码后,原子图和键图的表示共同参与分子性质预测。
结果
双图表示提升分子性质预测能力
研究人员首先对DGT进行了全面基准测试,共覆盖生理学、生物物理学、物理化学和量子化学四大领域的十个公开数据集。
在分类任务中,DGT在血脑屏障穿透、毒性预测、生物活性预测等任务上均取得最佳表现。相比现有图神经网络和预训练Transformer模型,其AUC进一步提高,说明双图表示能够更加准确地学习复杂分子结构与生物学性质之间的关系。
在回归任务中,DGT同样表现突出,无论是水溶性、脂溶性还是量子化学性质预测,误差均明显低于目前主流模型。尤其在HOMO和LUMO轨道能级预测任务中,其预测误差远低于其他图Transformer和三维图神经网络。
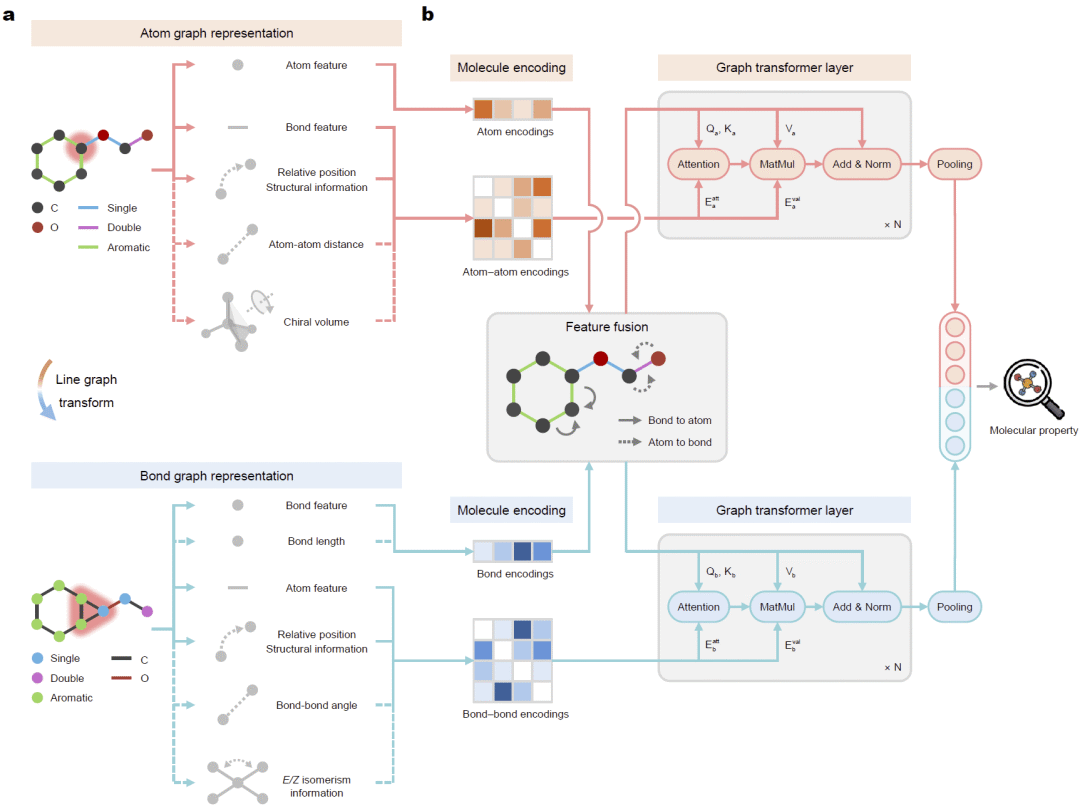
图1: 双图Transformer整体框架及原子图—键图表示。
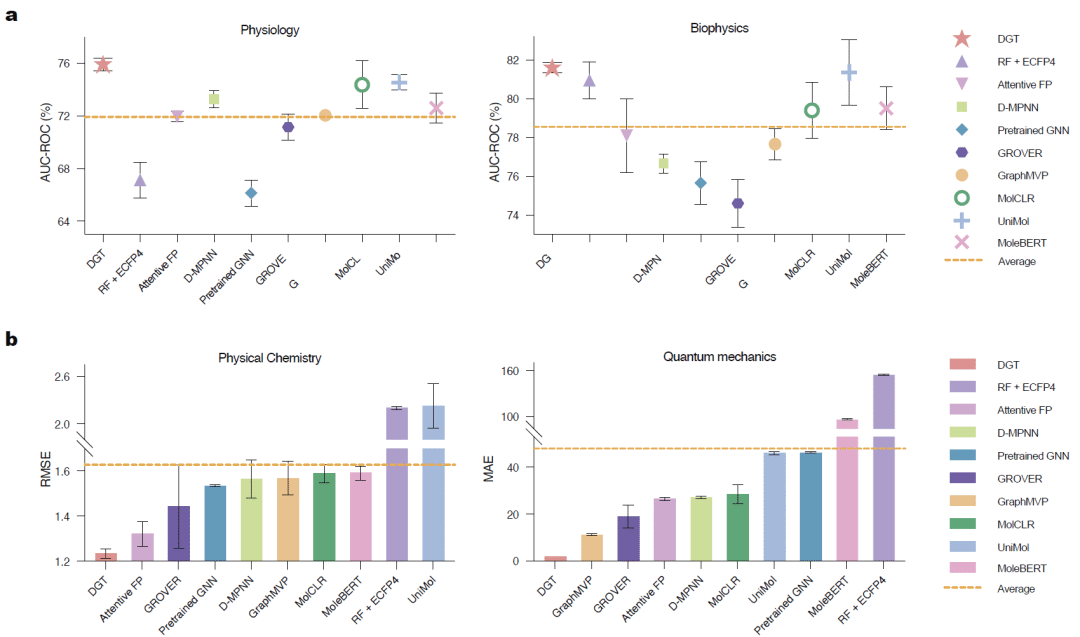
图2: DGT在四类分子性质预测任务上的性能比较。
双图表示和结构编码共同贡献模型性能
为了分析模型性能来源,研究人员分别移除不同模块进行消融实验。结果显示,仅保留原子图或仅保留键图都会导致预测性能下降,其中移除键图后预测误差增加约3%,移除原子图后误差增加约6%,说明两种表示方式能够互补提供不同层面的化学信息。
进一步去除最短路径编码、随机游走编码和环结构编码后,模型性能继续下降,尤其环结构信息对芳香环及杂环丰富分子的预测具有重要作用。这说明Transformer只有同时感知图拓扑、结构关系和节点特征,才能充分发挥自注意力机制优势。
三维结构和立体化学进一步提升预测能力
研究人员进一步考察三维几何信息对模型性能的影响。实验分别采用高精度量子化学优化结构以及MMFF、UFF等经典力场生成的近似构象。结果表明,引入三维几何信息后,HOMO和LUMO预测误差进一步降低,其中采用高精度三维结构时提升最明显,而近似构象同样能够带来稳定收益。
随后,研究人员将R/S手性和E/Z异构信息加入模型,在芳香化酶和雌激素受体毒性预测任务中,分类准确率进一步提高。结合立体异构数据进行预训练后,模型对对映异构体识别能力继续增强,说明立体化学信息对于药物相关任务具有重要价值。
这些实验表明,双图表示不仅能够融合二维拓扑信息,还能够自然整合三维空间和立体化学信息,实现更加完整的分子表示学习。
注意力机制揭示关键分子结构
除了预测性能,研究人员还分析了模型的可解释性。通过结合注意力权重和梯度信息,模型能够自动识别影响预测结果的重要原子和化学键。在血脑屏障预测任务中,模型重点关注氢键受体和杂环结构,这与经典药物设计规律一致。在BACE和HIV抑制剂预测中,注意力主要集中于芳香环、氢键位点和具有空间构型的关键官能团。
对于水溶性和脂溶性预测,模型重点关注极性基团和疏水表面;在量子化学性质预测中,引入三维信息后,模型更加准确地定位影响轨道能级的重要环结构。
研究人员还分析了两种典型立体异构药物——Fumagillin和Tamoxifen。结果显示,模型能够自动识别影响酶结合和受体结合的关键手性中心及E/Z构型,与实验结构生物学研究结果高度一致,证明模型具有良好的结构可解释性。
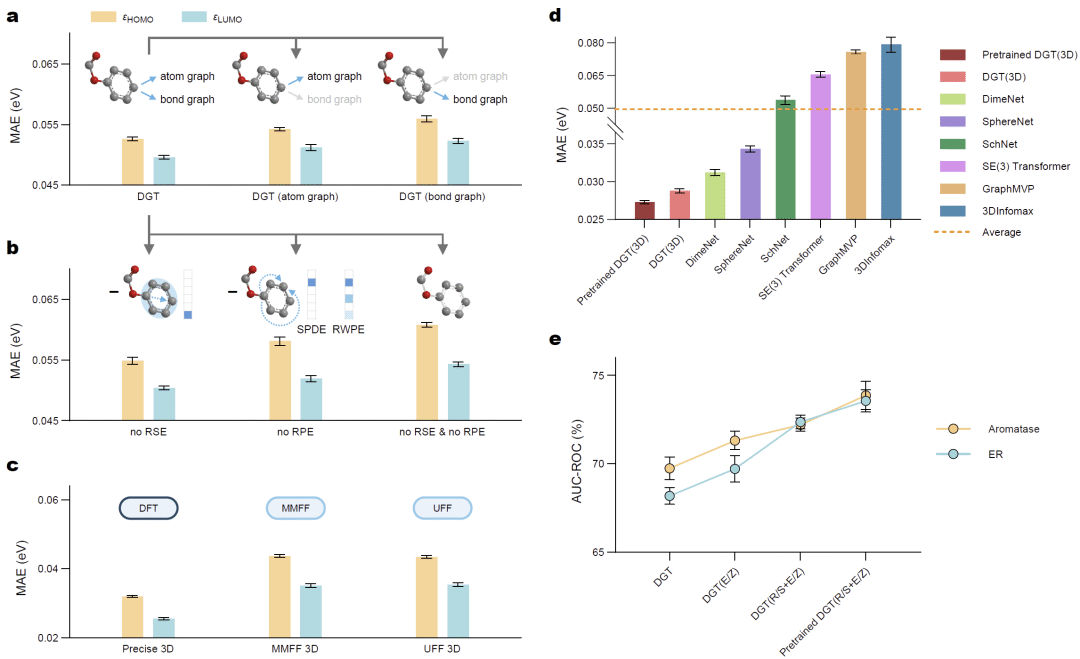
图3: 双图表示、三维信息及立体化学信息对模型性能的贡献分析。
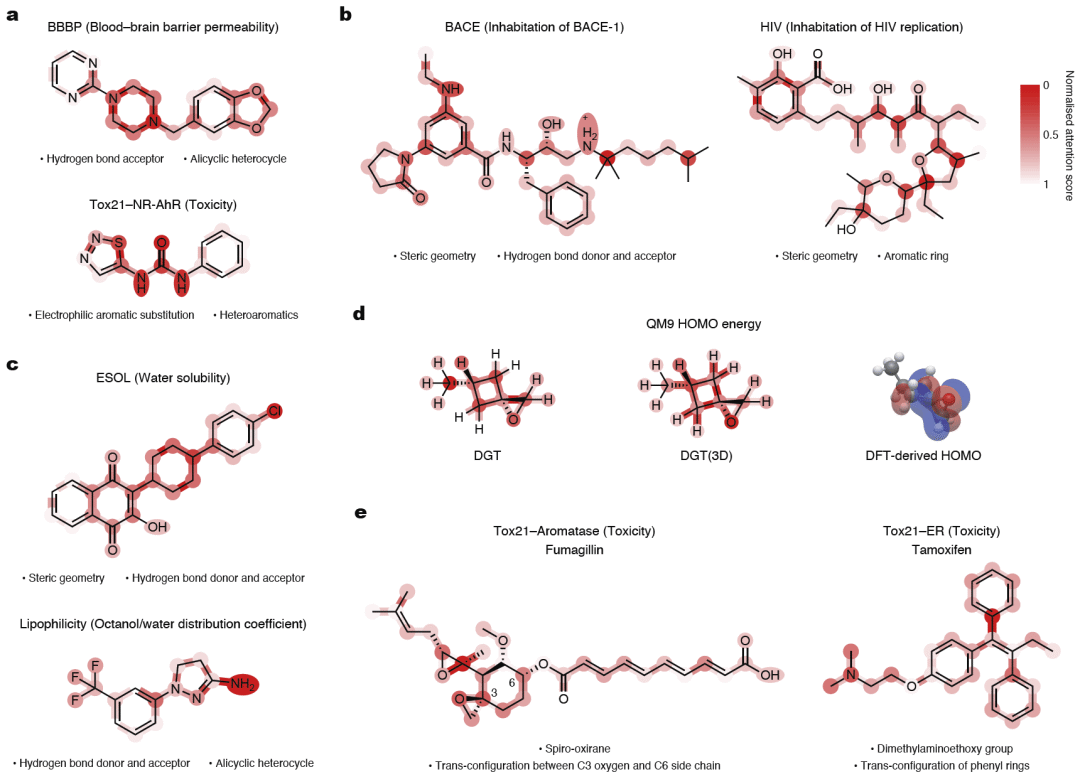
图4: 基于注意力机制的关键分子结构解释结果。
讨论
研究人员提出的Dual Graph Transformer为Transformer分子学习提供了一种新的表示范式。相比传统仅依赖原子图的方法,双图表示同时建模原子和化学键,使自注意力能够更加全面地学习局部化学环境和全局拓扑关系,从而显著提升分子性质预测性能。
研究进一步证明,图结构编码、三维几何信息和立体化学信息能够协同增强模型表达能力,而注意力机制还能直接揭示影响预测的重要结构区域,提高模型的可解释性。这些特点使DGT不仅具有较高预测精度,也更加符合化学知识。
不过,研究人员也指出,双图表示引入了原子—原子和键—键两套关系矩阵,其计算复杂度随分子规模平方增长,对于大分子仍存在一定计算开销。未来可结合稀疏注意力、层次化Transformer以及模型并行策略进一步提高计算效率。同时,还可将DGT扩展到分子构象生成、分子生成设计、化学反应预测等更复杂任务,并融合更多拓扑描述符和结构知识,进一步提升模型的泛化能力。
总体而言,Dual Graph Transformer展示了Transformer在分子机器学习中的巨大潜力,为高通量分子筛选、药物发现和人工智能驱动的化学研究提供了一种兼具高精度和高可解释性的全新框架。
整理 | DrugOne团队
参考资料
Zhang, S., Lapkin, A.A. Enhancing molecular property prediction of transformer models with dual graph representation. Nat Commun (2026).
https://doi.org/10.1038/s41467-026-75005-9
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-07-10,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号