Eur. J. Med. Chem. | 从数十亿分子中找到那把钥匙:超大化学空间结构虚拟筛选的进展与陷阱

Eur. J. Med. Chem. | 从数十亿分子中找到那把钥匙:超大化学空间结构虚拟筛选的进展与陷阱

DrugIntel
发布于 2026-03-30 16:04:53
发布于 2026-03-30 16:04:53

原文:Structure-based virtual screening of ultra-large chemical spaces: Advances and pitfalls 作者:François Sindt, Didier Rognan* 单位:法国斯特拉斯堡大学 CNRS 实验室 (UMR7200) 期刊:European Journal of Medicinal Chemistry 305 (2026) 118576 DOI:10.1016/j.ejmech.2026.118576
写在前面
如果说二十世纪的药物发现是在一个小型图书馆里翻阅书目,那么当前的计算药物化学已进入了一个规模相当于整个互联网的数字宇宙。化学空间的边界正以指数级速度向外扩张——某些私有化合物库已编码超过 个分子,而公开可购买的按需合成空间也已突破万亿()量级。
这不是数量级上的简单叠加,而是药物发现哲学的根本性转变:我们不再受限于"货架上有什么",而是开始思考"化学原理允许什么"。
这篇由斯特拉斯堡大学 Sindt 与 Rognan 联合撰写、2026年发表于《Eur. J. Med. Chem.》的综述,系统梳理了面向超大化学空间的基于结构虚拟筛选(Structure-Based Virtual Screening, SBVS)的四大技术路线,以翔实的前瞻性实验数据为支撑,深入剖析了各方法的技术本质、适用边界与现存陷阱。全文共引用 144 篇文献,覆盖该领域 2019–2025 年的核心进展。
一、概念厘清:化学空间 ≠ 化合物库
在进入技术细节之前,作者首先明确界定了两个在文献中常被混用的概念,这是理解全文的基础。
1.1 化合物库(Compound Library)
指实物在架、可立即购买的分子集合。每个分子均有明确的结构、价格及供货信息,可被完整枚举并以注释形式存储。通常规模不超过 1500 万个分子,代表供应商包括 Enamine、Sigma-Aldrich、Molport 等。
1.2 化学空间(Chemical Space)
指以构建块(building block)目录编号与有机化学反应序列编码的虚拟分子集合。这些分子尚未被合成,但理论上可通过 2–3 步并行合成实现。化学空间的关键特征是:
- • 不可穷举性:完整枚举需要巨量计算资源,通常不可行
- • 高合成成功率:按需合成成功率约为 80%,交货周期数周
- • 规模超越传统库数个量级:已达 –
表 1:代表性按需化学空间规模
供应商 | 名称 | 规模 | 类型 |
|---|---|---|---|
GSK | GSK-XXL | 私有 | |
Merck | MASSIV | 私有 | |
BioSolveIT | KnowledgeSpace | 虚拟 | |
Enamine | xREAL | 商业 | |
Synple Chem | Synple Space | 商业 | |
Enamine | REAL Space | 商业 | |
WuXi | GalaXi | 商业 | |
NCI | SAVI-Space-2024 | 虚拟 | |
传统化合物库 | (各供应商) | ~ | 实物在架 |
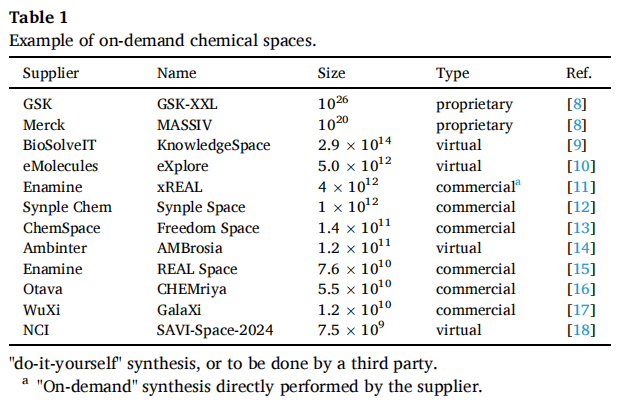
值得注意的是,各化学空间之间的重叠率极低,通常仅 0.2%–2%,这意味着它们彼此独立、互为正交的起点,为苗头发现和早期 SAR 建立提供了大量互补机会。
二、超大规模筛选的四大技术路线
文章将当前主流方法归纳为四条技术路线(见图 1 示意):
化学空间
│
├─── 穷举式对接 ──────────────→ DOCK3.7/3.8, VirtualFlow, ICM-Pro, AutoDock-GPU
│
├─── 机器学习加速对接
│ ├── 主动学习 ────────→ DeepDocking, MolPAL, RosettaVS, HASTEN
│ └── 被动学习 ────────→ Conformal Prediction, RAD
│
├─── 合成子驱动对接
│ ├── 合成子/骨架对接 → V-SYNTHES, Chemical Space Docking, SpaceDock
│ └── 迭代混合方法 ──→ HIDDEN GEM, SpaceHASTEN
│
└─── 进化算法 ────────────────→ SpaceGA, REvoLd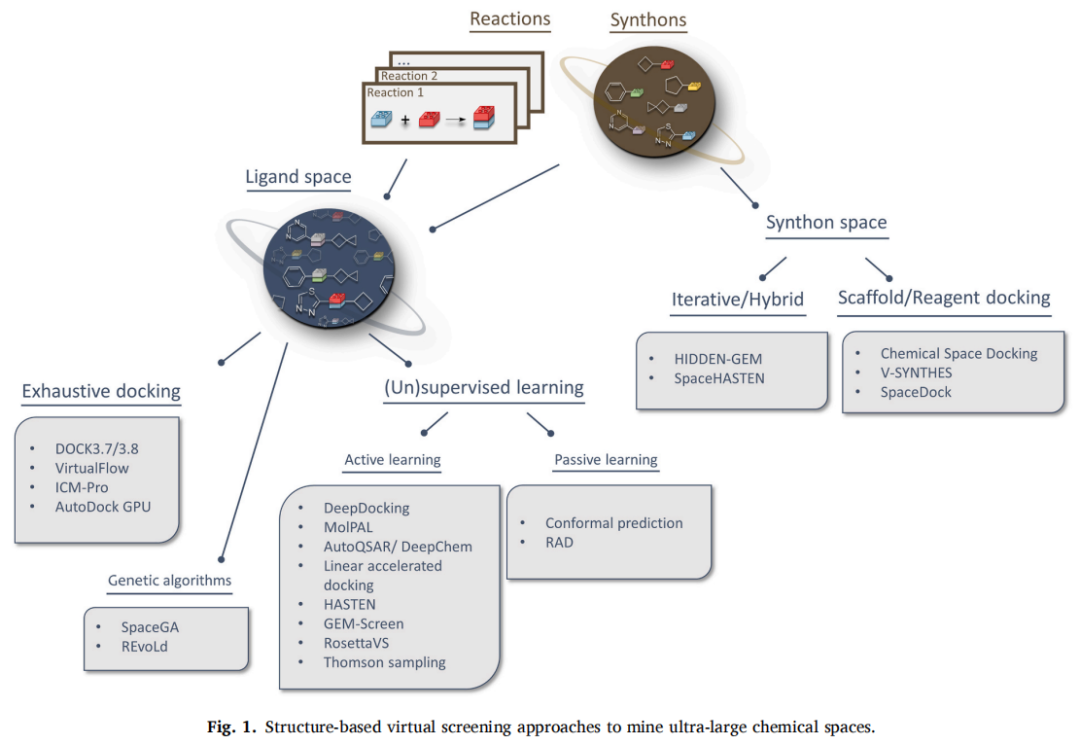
三、穷举式对接:奠基之战与规律总结
3.1 技术原理
穷举式对接(Exhaustive Docking)是最直接的超大规模筛选方式:逐一枚举全部待筛分子,将每个分子以标准对接算法放置于靶点结合口袋,输出打分排名。计算规模与化学空间大小呈线性关系,创新主要体现在两方面:一是高性能计算集群的并行化调度;二是对接引擎本身的架构优化(尤其是 GPU 加速)。
3.2 关键里程碑
2019 年:UCSF 奠基之战
Lyu、Irwin 与 Shoichet(UCSF)在 Nature 上发表了首个超大规模对接研究,确立了该领域的方法论范式:
- • 靶点:AmpC β-内酰胺酶 + 多巴胺 D4 受体(DRD4)
- • 规模:约 1 亿按需合成化合物
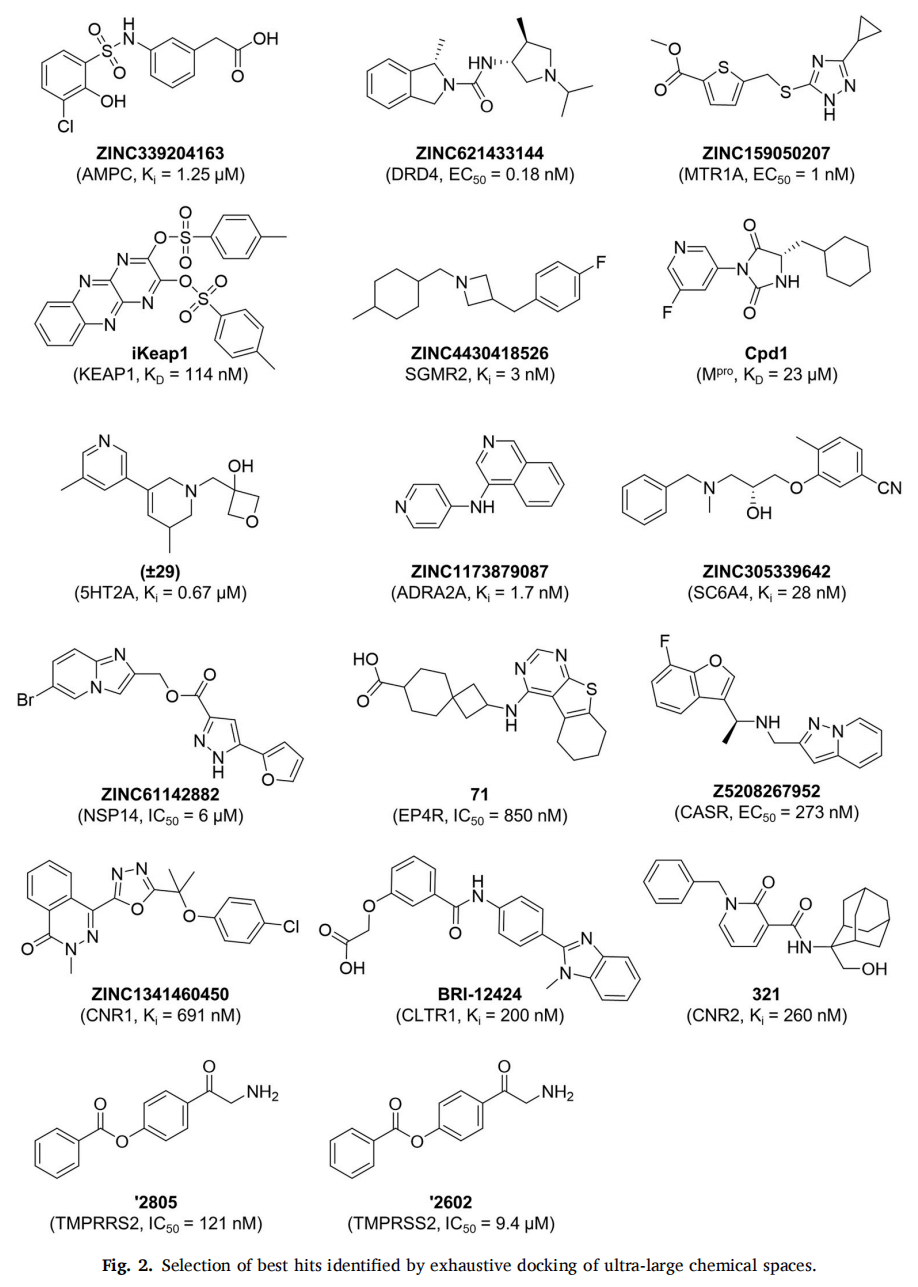
- • DRD4 命中率:22%(549 个测试化合物中 122 个有活性),获得多个纳摩尔配体,最优 EC₅₀ = 180 pM,亚型选择性超过 2500 倍
- • 重要发现:(i)对接打分随库规模增大而提升;(ii)筛选超大库可发现独特化学骨架;(iii)大多数苗头化合物在传统"现货"库中不存在;(iv)先按打分排序再人工视觉检查是最优策略
代表性前瞻应用汇总(DOCK 系列,截至 2025 年):
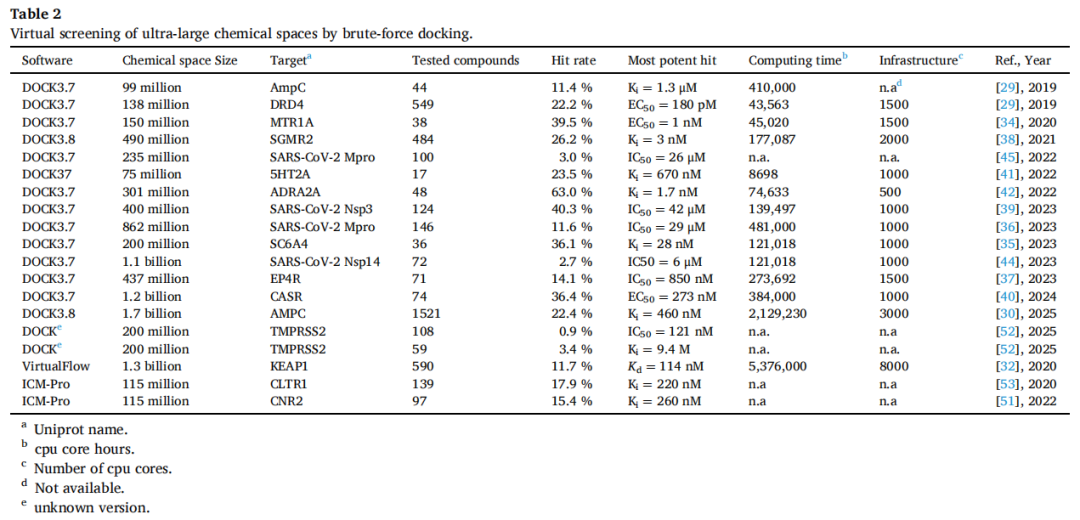
3.3 GPU 加速的突破
GPU 在架构上天然适合大规模并行计算,正在成为穷举对接的加速引擎:
- • Uni-Dock:相比单 CPU 核 AutoDock Vina 实现超过 1000 倍加速,对 KRAS^G12D^ 的 3820 万分子筛选在 100 块 NVIDIA V100 内 12 小时完成
- • AutoDock-GPU:在 Summit 超算(200 petaFLOPS,27,648 块 V100)上估计可在单日完成 10 亿分子对接
- • RIDGE:将对接引擎直接嵌入 GPU,最小化主机-设备数据传输,实现约 100 化合物/秒 的对接速度
3.4 穷举对接揭示的五大规律
作者总结了超大规模穷举对接累积五年来浮现的稳健规律:
规律一:对接打分随库规模增大而提升
无论使用何种对接工具或打分函数,筛选更大的化学空间均可获得更好的对接打分。逻辑上,更大的库包含更多能与蛋白结合口袋完美匹配的分子。
规律二:命中率随库规模增大而提升
以 AmpC β-内酰胺酶为例(同一靶点、两次不同规模筛选的直接对比):
- • 9900 万分子:命中率 11%,最优抑制剂 Ki = 1.3 μM
- • 17 亿分子:命中率 22%,最优抑制剂 Ki = 460 nM
对钙敏感受体(CaSR)的研究显示,大规模筛选(12 亿)命中率是小规模(270 万)的近 3 倍,平均活性强 37 倍。
⚠️ 陷阱提示:化合物数量的增加同样带来了假阳性(artifacts)的增加。这些假阳性化合物通常含有高极性基团,被大多数基于经验力场的打分函数错误地赋予高分,可通过结构过滤和溶剂化能计算加以排除。
规律三:命中率随对接打分提升而增大
传统观点认为对接打分与结合自由能几乎不相关,但超大规模对接揭示了一个微妙的统计规律:打分分布与活性分布在统计层面存在对应关系。以 17 亿化合物的 AmpC 筛选为例,在不同打分区间选取化合物进行测试,命中率随打分提升而清晰递增——这部分挑战了"打分完全无效"的传统论断。
规律四:实验测试规模影响命中率的稳定性
当化学空间扩展至数十亿量级时,潜在命中化合物的数量也将激增(例如 DRD4 在 1.38 亿化合物中的预估活性配体约 45.3 万个)。若仍沿用传统的"购买50–100个化合物测试"策略,命中率将高度依赖随机抽样的运气。作者建议:对成药性好的靶点(GPCRs、激酶)至少选择 150–200 个化合物测试,对难成药靶点应达到 500 个。
规律五:超大规模筛选苗头具有独特生物学特性
这是该领域最令人瞩目的发现之一:超大化学空间筛选发现的苗头化合物,往往展现出传统筛选难以获得的特殊药理学性质:
靶点 | 特殊性质 |
|---|---|
DRD4 全激动剂(EC₅₀ = 180 pM) | 亚型选择性超过 2500 倍 |
MTR1A 选择性激动剂 | 黄昏时给药使小鼠昼夜节律提前 1.5 小时 |
5-HT₂A 激动剂 | 具有抗抑郁特性,无致幻活性,药代动力学异常 |
α₂A 肾上腺素受体激动剂 | 无镇静效应的非阿片类镇痛剂 |
血清素转运体(SC6A4)抑制剂 | 稳定外向-关闭构象,具强效抗抑郁/抗焦虑活性 |
EP4R 拮抗剂 | 外周限制性,强效抗异常疼痛和抗炎 |
CaSR 正变构调节剂 | 降低甲状旁腺素而不诱发低钙血症 |
CLTR2^L129Q^ 逆激动剂 | 为葡萄膜黑色素瘤提供新治疗策略 |
作者坦承,目前尚需更多案例研究来建立"库规模"与"生物学独特性"之间的直接因果关系,但超大库中绝大多数化合物在传统库中不存在(75% 的已验证苗头不可在 4300 万"现货"化合物中购得)这一事实,至少支持了此种关联的合理性。
3.5 穷举对接的极限
不同规模穷举对接的计算需求估算
规模 | 化合物数量 | CPU 核时 | 估算耗时 | 存储需求 | 估算费用(USD) | 可行性 |
|---|---|---|---|---|---|---|
小型 | (1000万) | 5,500 | ~5 小时 | ~100 GB | $230 | 高 |
超大型 | (10亿) | 550,000 | ~23 天 | ~10 TB | $23,000 | 可行 |
万亿级 | (1万亿) | 5.5 亿 | ~63 年 | ~10 PB | $2300 万 | 不可行 |
注:基于平均 2 秒/化合物的对接速度,1000 核 CPU 基础设施,标准云计算价格(AWS/Google Cloud)估算
此外,仅分子准备阶段(2D→3D转换、质子化态计算、构象生成)就极为耗时:以 Schrödinger GlideHTVS 处理 15.6 亿分子为例,需要约 30 天计算,消耗约 457,600 核时——约为对接步骤本身的 1/3 至 1/2。
结论:穷举式对接的实际上限约为 10–20 亿分子。
四、机器学习加速对接:效率的突破与现实的落差
4.1 核心思路
ML 加速对接的基本逻辑是以小博大:仅对 1%–5% 的子集实施实际对接,以此为训练数据,构建 ML 模型预测全库打分,从而将大量计算资源集中于最有前途的候选分子。
从模型输出类型分为:
- • 分类器(Classifier):以预定义打分阈值将分子分为"苗头"与"非苗头"
- • 回归器(Regressor):直接预测对接打分的具体数值
从学习策略分为:
- • 主动学习(Active Learning, AL):迭代式反馈循环——每轮以采集策略(如贪婪、UCB、Thompson 采样)选择新分子实际对接,更新模型,直至收敛
- • 被动学习(Passive Learning):一次性训练,无需反馈循环
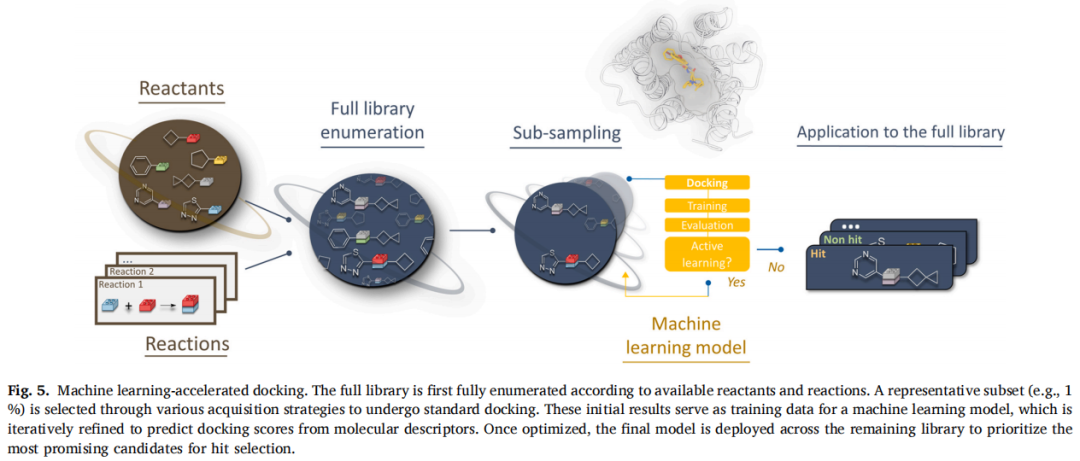
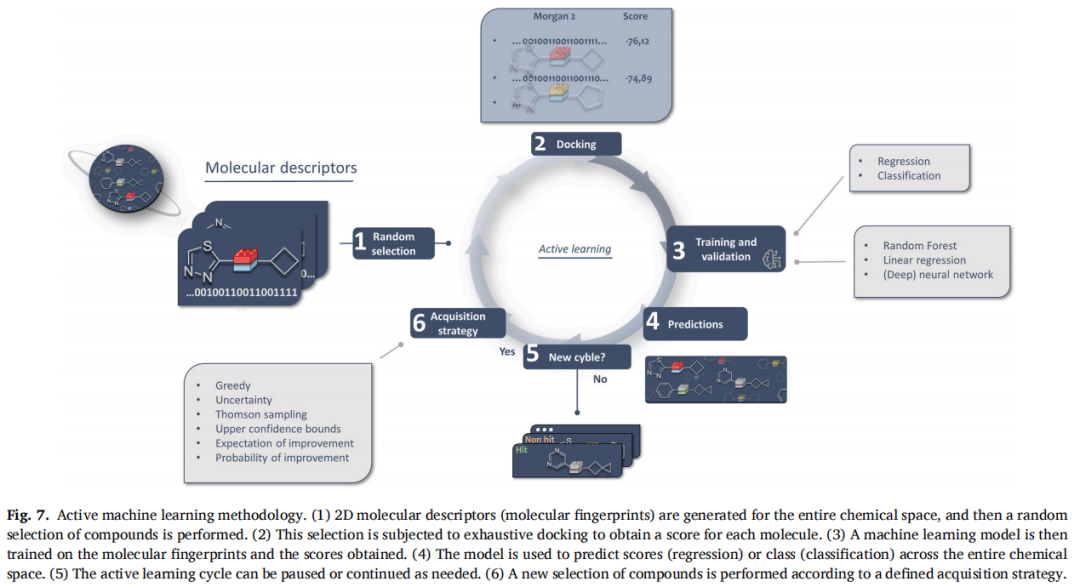
4.2 代表性前瞻应用
(1)DeepDocking(DD)
目前应用最广、验证最充分的 ML 加速对接方法之一,由 Cherkasov 课题组(UBC)开发。
- • 架构:深度神经网络(DNN)+ 主动学习 + Morgan-2 指纹
- • 工作流程:随机子集对接 → DNN 训练 → 全库预测 → 主动学习循环(重复5–12轮)→ 最终虚拟苗头穷举对接
- • 代表应用:
- • 400 亿分子筛选 SARS-CoV-2 Mpro(融合 ZINC15 + REAL Space),仅对 0.45% 的分子实际对接,命中率 9.1%,最优 IC₅₀ = 10.8 μM;使用了 640 CPUs + 250 GPUs
- • Aβ42 纤维筛选(5.3 亿分子),命中率高达 54%,最优两个苗头 KD = 7 nM 和 13 nM
(2)RosettaVS
由 DiMaio 课题组(UW)开发,基于 Rosetta 力场的前馈神经网络(FFNN)方法。
- • 技术特点:以 2D 圆形指纹预测 RosettaGenFF-VS 对接打分,主动学习循环最多 10 轮,最终入选化合物以考虑蛋白柔性的 VSH 模式重新对接
- • 代表应用(KLHDC2 泛素连接酶):发现全球首批 KLHDC2 已知配体,命中率 14%,最优 IC₅₀ = 2.9 μM,对接构象经 X 射线晶体衍射验证;仅需 0.11% 化学空间显式对接,3000 CPUs + 1 GPU,不足一周完成
(3)Conformal Prediction(CP)
由 Luttens 等(Carlsson 课题组)提出,与标准 ML 不同之处在于可量化每个预测的置信区间。
- • 以 CatBoost(梯度提升决策树)为基础模型,引入可调节误差阈值参数
- • 在 DRD2 筛选中:3.5 亿化合物空间,仅训练于 100 万化合物(0.42%),梯度提升模型筛出 2400 万虚拟苗头;最终测试 31 个化合物,两个命中(Ki = 3–4 μM)
- • 双靶点筛选(A2AR/DRD2):找到 1 个具有双重活性的分子(Ki(DRD2) = 14 μM,Ki(A2AR) = 20 μM)
4.3 代表性回顾性研究
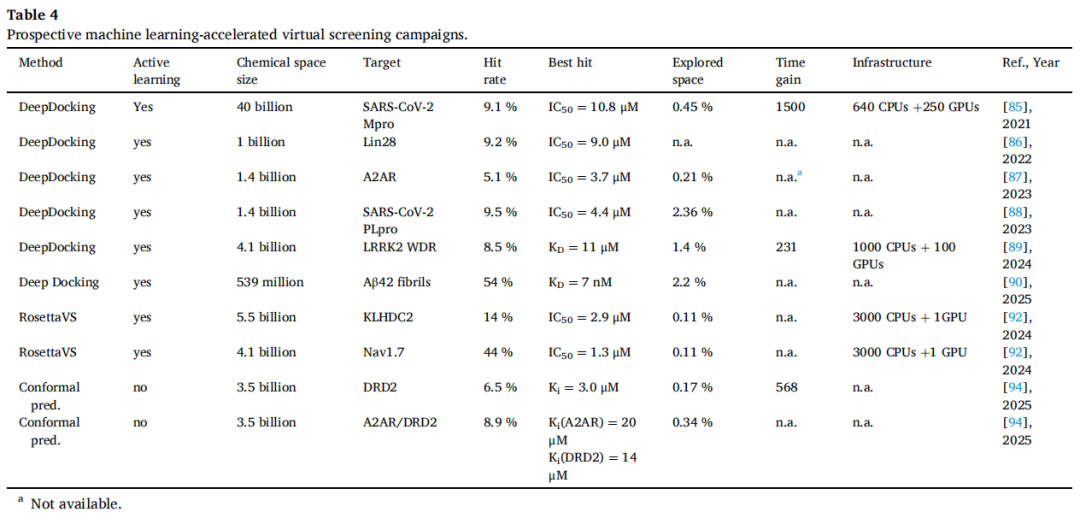
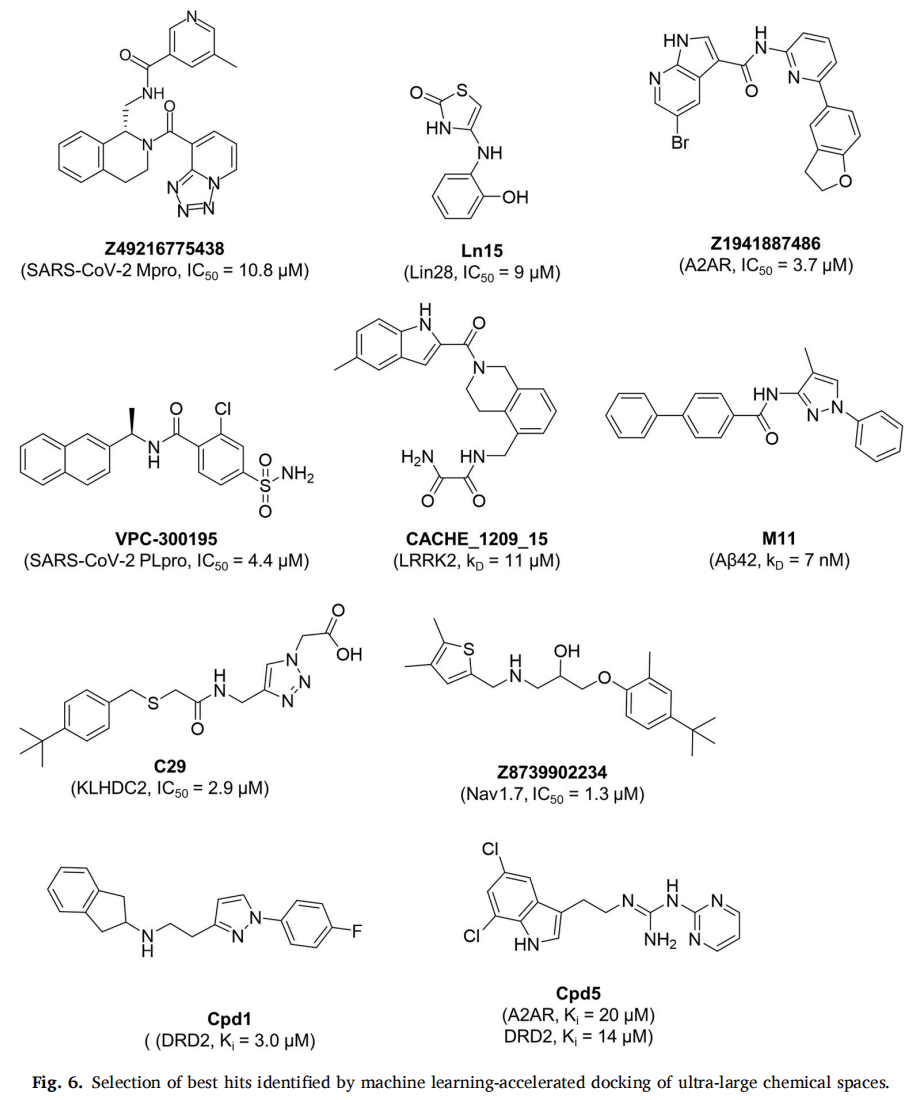
大量回顾性研究以 AmpC β-内酰胺酶(9900 万,DOCK3.7)和 DRD4(1.38 亿,DOCK3.7)数据集为基准测试,主要结论如下:
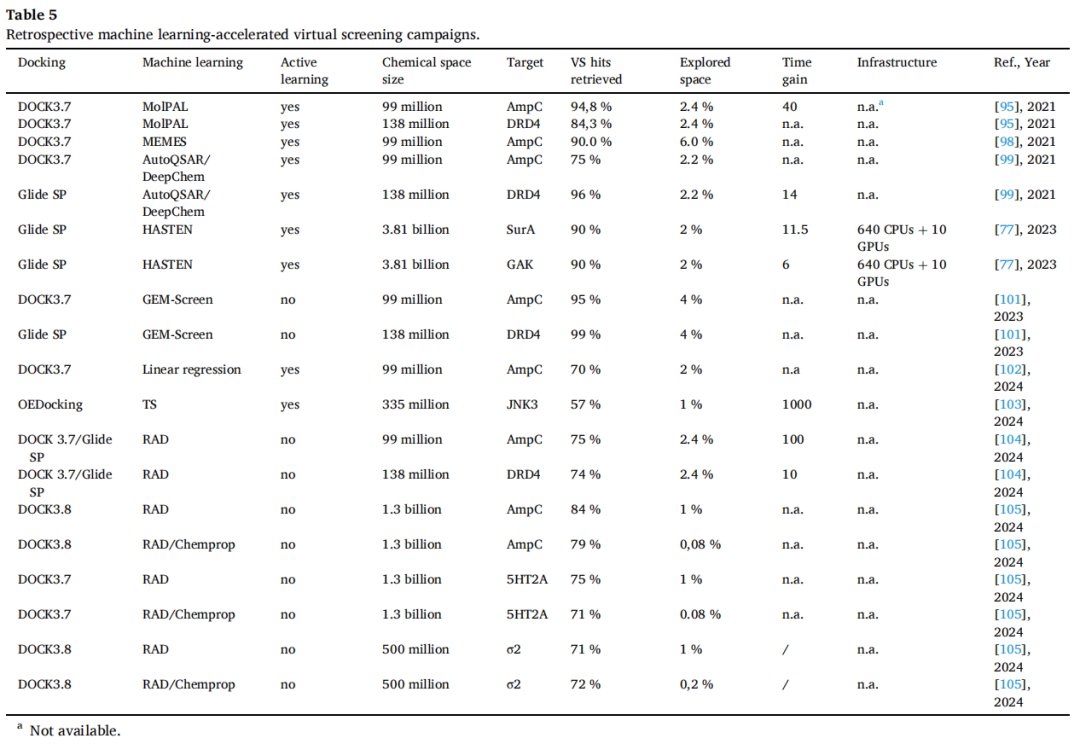
方法 | 架构 | AmpC 虚拟苗头回收率 | DRD4 虚拟苗头回收率 | 探索空间比例 |
|---|---|---|---|---|
MolPAL (D-MPNN + UCB) | 图神经网络 | 94.8% | 84.3% | 2.4% |
AutoQSAR/DeepChem (GCNN) | 图卷积网络 | 75% | 96% | 2.2% |
HASTEN (D-MPNN + 贪婪) | 图神经网络 | 90% | 90% | 2% |
GEM-Screen (3D GNN) | 三维图神经网络 | 95% | 99% | 4% |
MEMES (Bayesian GP) | 贝叶斯优化 | 90% | — | 6% |
RAD (HNSW 图) | 近邻检索 | 75% | 74% | 2.4% |
线性回归 (LR) | 线性模型 | 70% | — | 2% |
Thompson Sampling | 概率搜索 | 57%(top100) | — | 1% |
值得注意:线性回归(LR)以几乎最快的训练速度(比非线性模型快约 100 倍)达到 70% 回收率,接近复杂深度学习方法,体现了"复杂度不总是转化为更好效果"的重要提示。
4.4 ML 加速对接的深层反思
作者对该领域的现状做出了较为犀利的评价,值得重点关注:
问题一:高相关系数不等于高质量预测
大多数 ML 模型以 Pearson 相关系数(Rp)评估,通常在 0.70–0.80 之间,看似可观。但模型往往倾向于学习分数分布的整体趋势而忽视最顶部打分的化合物——而这恰恰是虚拟筛选最关心的。一项研究显示:模型在 150 亿化合物上达到 Rp = 0.83,但仅能优先化 13.5% 的实验活性化合物。
问题二:ML 模型精度上限受原始对接引擎制约
ML 所模拟的对接打分本身对结合自由能的预测精度就有限,因此 ML 对接的性能天花板由其模拟的对接工具决定——好的模型只能尽量逼近对接的结果,而无法超越对接本身的精度。
问题三:前瞻验证案例严重匮乏
迄今仅有 3 种方法(DeepDocking、RosettaVS、Conformal Prediction) 完成了真正的前瞻性实验验证,绝大多数方法仅停留在回顾性基准测试阶段。已验证的前瞻应用中,命中率和苗头活性普遍低于穷举对接(通常命中率 5%–10% vs. 穷举的 10%–40%;最优苗头多为双位数微摩尔 vs. 穷举的纳摩尔至亚微摩尔)。
问题四:万亿级空间使 ML 同样陷入困境
若化学空间达到 ,即使训练集限于 0.1%,也需要对 10 亿化合物进行实际对接——计算成本极为高昂。更根本的是,对万亿化合物进行全库预测推断本身就几乎不可行。
五、合成子驱动组合对接:最具前景的范式创新
5.1 核心思想
合成子方法从根本上与穷举/ML 方法不同——它不从产品空间出发,而是直接在试剂(reagent)或合成子(synthon)空间操作。其逻辑与按需化学空间的构建逻辑天然契合:化学空间本由试剂+反应定义,那么直接在这一层面探索即可,无需枚举全部产物。
这是目前唯一被证实可处理万亿级化学空间的结构虚拟筛选策略,也是文章作者重点推介的方向。
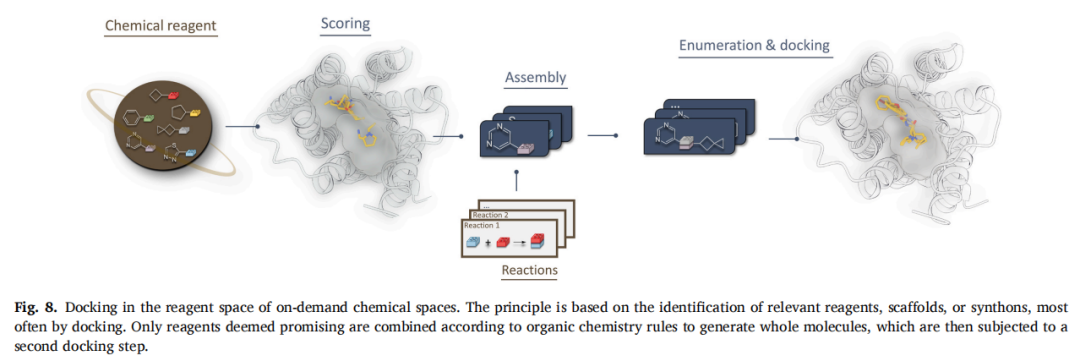
5.2 三大代表方法的技术细节
(1)V-SYNTHES / V-SYNTHES2(Katritch 课题组,USC)
- • 核心机制:为每个反应类型定义 Markush 结构(公共骨架 + R 基),将合成子转化为最小枚举库(MEL,以甲基或苯基替代),对约 60 万 MEL 实施 ICM-Pro 对接,选出高打分的 MEL 后移除帽基,再与所有相容合成子组合形成约 100 万化合物的子库进行第二轮对接
- • 效率:相比穷举对接节省约 5000 倍 计算量
- • 前瞻验证(CNR1/CNR2):靶向 110 亿化合物空间,仅需 210 万次对接,命中率 33%(两受体均),最优配体 Ki = 280 nM(CNR1),随后从 REAL Space 筛出 23 个纳摩尔优化分子,其中一个 Ki = 0.9 nM(CNR2,50 倍对 CNR1 选择性)
- • V-SYNTHES2:扩展至 360 亿化合物空间,全自动 pose 筛选,仅需 40,000 核时、约 300 美元成本
(2)Chemical Space Docking(CSD)(BioSolveIT)
- • 核心机制:使用 72,000 个商业构建块,以伪原子替代活性基团定义"合成子"(与 V-SYNTHES 不同),在药效团约束下以 FlexX + HYDE 打分函数对合成子对接,选出前 500 个合成子,每个合成子生成约 1 万个化合物子库进行模板匹配对接
- • 前瞻验证(ROCK1):测试 69 个化合物,命中率 39%,最优 Ki = 38 nM,13 个亚微摩尔苗头
- • 前瞻验证(PKA):以4个晶体片段为约束,探索 26 亿化合物空间,命中率 40%(30/93),最优 Ki = 744 nM;已商业化
(3)SpaceDock(Sindt & Rognan,斯特拉斯堡,本文作者之一的原创工作)
这也是综述作者自己开发的方法,在文章中有较为详细的描述,具有以下独特性:
- • 使用未修改的商业构建块直接对接(无需帽基替换),对每个反应物生成 20 个构象并对接,按拓扑和化学相容性规则组合,快速能量最小化后重新对接验证
- • 引入姿态质量检查(排除扭曲构象、高应变能、过多未满足 H 键的分子)
- • 对接+组合成功率极低(双组分反应约 ,三组分约 ),但已通过主动学习(SpaceDock + ML 预测试剂对兼容性)加速,可用 5% 的化学空间探索恢复 98% 的虚拟苗头
- • 前瞻验证(DRD3):670 亿化合物空间,命中率 60%(6/15),多个亚微摩尔苗头,最优 Ki = 320 nM,全新骨架
- • 前瞻验证(CHI3L1):377 万亿化合物空间,2.6 亿化合物被优先化重对接,45 个化合物测试,命中 3 个,最优 Kd = 19 μM(SPR 正交验证),6 周完成从筛选到实验的全流程
5.3 混合方法
(4)HIDDEN GEM(主动学习 + 生成模型 + 相似性搜索)
- • 工作流:小型多样性库对接 → 生成模型(以 ChEMBL 活性化合物训练)生成 de novo 结构 → 与 Enamine REAL(370 亿化合物)进行 2D 相似性搜索检索可购买类似物 → 重新对接,闭合主动学习循环
- • 对 16 个靶点展示了苗头打分提升,但尚无前瞻性实验验证,且 2D 相似性矩阵计算需要对全库枚举,计算代价较大
(5)SpaceHASTEN(ML + 化学空间相似性搜索)
- • 工作流:GNN 预测 100 万多样性种子分子的 Glide SP 打分 → 以 SpaceLight 和 FTrees 两个超快工具在未枚举的 480 亿化合物 REAL Space 中检索类似物 → 贪婪采集更新 top-1000,循环两次,共约 300 万次对接
- • 对 3 个靶点(KIF11、TGFR1、PYRD)展示了筛出高打分化合物的能力,但同样尚无前瞻实验验证
5.4 进化算法
(6)SpaceGA(Moesgaard & Kongsted)
- • 以随机池(1 万化合物)初始化,对接打分为适应度函数;突变/交叉仅在购买空间内有同类似物时才允许,通过 SpaceLight 2D 拓扑相似性保证可购买性
- • 约对接 100 万化合物后可生成约 30% 直接可购买的化合物,但化学多样性有限(倾向利用已知优解)
(7)REvoLd(Meiler 课题组,Rosetta 遗传算法)
- • 直接在试剂+反应空间进行进化,从 200 个随机配体出发,每轮仅生成 50 个新个体(偏重骨架探索的交叉+单试剂突变),约 30 代后收敛
- • 每靶点约 5–7 万次对接,1–2 天完成(<100 CPU 核);在 CACHE 国际挑战 #1 中成功发现 3 个 LRRK2 WDR 弱抑制剂
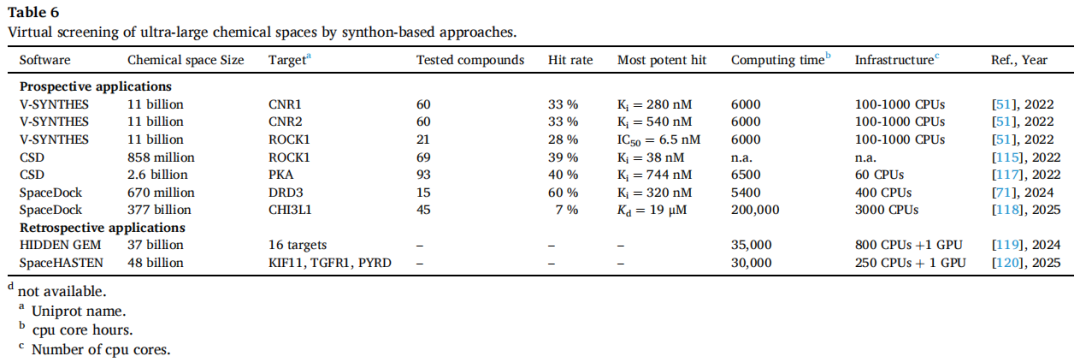
5.5 合成子方法的关键优势与现存局限
优势:
- • 与按需化学空间的底层逻辑(试剂+反应)完全契合,天然适配万亿级规模
- • 计算资源需求合理,数百核 + 1 TB 磁盘、不超过一周可完成
- • 命中率和苗头活性与穷举对接相当
- • V-SYNTHES 和 CSD 已商业化,存在大量未发表应用
局限:
- • 仍依赖对接的固有局限(打分函数精度、蛋白柔性处理等)
- • 对成药性差的结合口袋(如 CHI3L1)命中率显著下降
- • 万亿级三组分反应空间的方法验证尚不充分
- • 部分混合方法(HIDDEN GEM、SpaceHASTEN)缺乏前瞻实验验证
六、方法论横向比较
四大技术路线综合比较
维度 | 穷举对接 | ML 加速对接 | 合成子方法 | 进化算法 |
|---|---|---|---|---|
最大可处理规模 | ~10–20 亿 | ~400 亿 | 万亿级 | 万亿级(理论) |
是否需要全库枚举 | 是 | 是(预测阶段) | 否 | 否 |
前瞻验证充分性 | ★★★★★ | ★★☆☆☆ | ★★★★☆ | ★★☆☆☆ |
命中率(已验证) | 10–40% | 5–10% | 7–60% | 待验证 |
最优苗头活性 | 纳摩尔级 | 双位数微摩尔 | 纳摩尔至微摩尔 | 待验证 |
计算成本 | 高 | 中 | 低–中 | 低 |
可购买性保证 | 需后处理 | 需后处理 | 天然内置 | SpaceGA/REvoLd 内置 |
化学多样性 | 高 | 中(取决于采集策略) | 高 | 偏低(SpaceGA) |
工业化成熟度 | 高 | 中 | 高(CSD、V-SYNTHES 已商业化) | 低 |
七、当前挑战与未来展望
7.1 已识别的核心挑战
挑战一:假阳性随库规模增大而增多
高打分列表中假阳性分子的比例随库规模扩大而上升,通常具有扭曲构象或被经验打分函数优化的极性基团。解决方案包括:人工视觉检查(移除扭曲分子)、配体应变能过滤、溶剂化能计算(MM-GBSA 等)。
挑战二:对接打分仍然是糟糕的结合自由能代理
这是整个基于结构虚拟筛选领域的本质瓶颈,不因库规模变化而消失。提升后处理重打分的精度(包括物理化学方法和 AI 方法如 Boltz-2)是一个重要的改进方向。
挑战三:靶点结构输入的质量与来源
目前几乎所有案例均依赖高分辨率实验结构(X 射线、冷冻电镜)。AlphaFold 等 AI 预测结构是否可替代实验结构用于超大规模筛选?现有唯一报道显示,冷冻电镜和 AlphaFold2 结构可发现不重叠的配体集合、命中率相近,初步支持 AI 结构的适用性,但系统研究仍缺乏。
挑战四:核酸靶点的扩展
目前所有应用均聚焦于蛋白靶点。核酸(DNA/RNA)结合位点的独特性(更大、更具柔性、水介导相互作用更多)使得直接将现有方法迁移存在相当挑战,需要专门开发和验证。
7.2 作者的未来展望
近期(1–3 年内):
- • 开放科学挑战赛(如 CACHE 系列)将提供标准化基准,帮助客观评估 ML 加速对接与合成子方法的真实前瞻性能
- • 合成子方法向更多难成药靶点的扩展,更好地界定适用边界
- • GPU 算力的持续提升将推动穷举对接的可处理规模上限突破
中远期(3–5 年):
- • AI 对接方法(流匹配、扩散模型k)有望成为物理对接的替代方案,实现基于蛋白口袋条件的生成式配体设计
- • 超大化学空间的探索可能不再依赖单一的"筛选→购买→测试"范式,而是与自动化并行合成、快速 SAR 建立深度整合
- • 万亿级化学空间的优势(能否显著提升命中质量)尚待前瞻验证证实
八、编者按:为何此文值得精读?
这篇综述的学术价值体现在三个层面:
第一,系统性与批判性的统一。 文章不是简单的文献堆砌,而是在系统梳理四大方法体系的基础上,对每条路线的本质局限做出清晰而诚实的评价——尤其是对 ML 加速对接前瞻验证不足问题的直接点名,体现了作者超越了自身领域的客观视角。
第二,数据驱动,论据充分。 文章依托超过 20 个具有实验验证的前瞻筛选案例,以及数十个回顾性基准研究,为每一个方法论结论提供了坚实的数据基础,而非依赖理论推演。
第三,作者本身即领域核心贡献者。 Sindt 与 Rognan 不仅是综述者,SpaceDock 即为 Sindt 的原创工作,他们在 CHI3L1、DRD3 等靶点上的前瞻应用也列于文中。这意味着文章具有相当程度的一手经验与内部视角。
对于计算药物化学研究者而言,这篇综述提供了清晰的方法选择路线图;对于实验药化学家,它揭示了超大化学空间如何从根本上改变了苗头发现的逻辑;对于药物发现决策者,它量化了不同规模筛选的成本-效益边界。
参考文献(重点引用)
- 1. Lyu J. et al. Nature 566, 224–229 (2019). — 超大规模对接奠基之作
- 2. Liu F. et al. Nat. Chem. Biol. 21, 1039–1045 (2025). — 17亿化合物AmpC筛选
- 3. Gorgulla C. et al. Nature 580, 663–668 (2020). — VirtualFlow 平台
- 4. Sadybekov A.A. et al. Nature 601, 452–459 (2022). — V-SYNTHES
- 5. Beroza P. et al. Nat. Commun. 13, 6447 (2022). — Chemical Space Docking
- 6. Sindt F. et al. ACS Cent. Sci. 10, 615–627 (2024). — SpaceDock
- 7. Gentile F. et al. Chem. Sci. 12, 15960–15974 (2021). — DeepDocking, 400亿分子
- 8. Zhou G. et al. Nat. Commun. 15, 7761 (2024). — RosettaVS
- 9. Luttens A. et al. Nat. Comput. Sci. 5, 301–312 (2025). — Conformal Prediction
- 10. Graff D.E. et al. Chem. Sci. 12, 7866–7881 (2021). — MolPAL
- 11. Sindt F. et al. J. Chem. Inf. Model. 65, 5553–5566 (2025). — 超大对接苗头的重打分难题
- 12. Ackloo S. et al. Nat. Rev. Chem. 6, 287–295 (2022). — CACHE 挑战赛
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-22,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号