小分子药物发现预测建模的大挑战:一份来自一线药物猎手的技术清单

小分子药物发现预测建模的大挑战:一份来自一线药物猎手的技术清单

DrugIntel
发布于 2026-03-30 16:08:36
发布于 2026-03-30 16:08:36

论文信息 标题:Grand Challenges for Predictive Modeling in Small Molecule Drug Discovery 通讯作者:Woody Sherman(PsiThera, Watertown, MA) 参与机构:MIT · 耶鲁大学 · 意大利技术研究所 · 格罗宁根大学 · 莱顿大学 · 乌普萨拉大学 · Chemical Computing Group 等,共 14 位作者 数据来源引用截止:2026 年 3 月(Dimensions AI)
目录
- 1. 背景与动机:为什么此时需要一份 大挑战 清单
- 2. 论文框架与方法论
- 3. 化学领域(Chemistry):从分子设计到实物合成
- 4. 结构领域(Structure):从序列到三维空间
- 5. 能量领域(Energy):分子识别的热力学与动力学
- 6. 药理学领域(Pharmacology):从分子到体内行为
- 7. 横切主题与系统性反思
- 8. 对不同受众的启示
- 9. 总结评价
一、背景与动机
1.1 一个反直觉的现象
过去十年,AI 在蛋白质结构预测(AlphaFold)、分子生成(generative models)等方向取得了轰动性突破。然而,药物发现的整体效率并未随之发生可量化的跃升:新药研发的平均成本仍在数十亿美元量级,临床失败率依然高居不下,从靶点发现到上市的周期也没有实质性缩短。
作者们对这一现象的解释颇具洞察力:问题不仅在于方法的准确性不足,更在于 "我们连要解决什么问题都没说清楚"。当一个领域缺乏清晰定义的挑战目标和可量化评估标准时,计算创新的惯性就会偏向 在 benchmark 上刷分 ,而非解决真实项目中的决策瓶颈。
1.2 计算方法的历史演进
论文通过 Dimensions AI 数据库对 2010 年以来主要计算方法的引用量进行了系统追踪(图 1),揭示了一个清晰的规律:
时代 | 代表方法 | 热潮特征 |
|---|---|---|
2010–2014 | QSAR、分子对接、虚拟筛选、药效团模型 | 规则驱动,专家系统为主 |
2014–2018 | 分子动力学、自由能方法、QM/MM | 物理模型精度提升 |
2018–2022 | 深度学习、生成模型、强化学习 | 数据驱动爆发 |
2020 至今 | AlphaFold、共折叠、蛋白-配体生成 | 结构预测革命 |
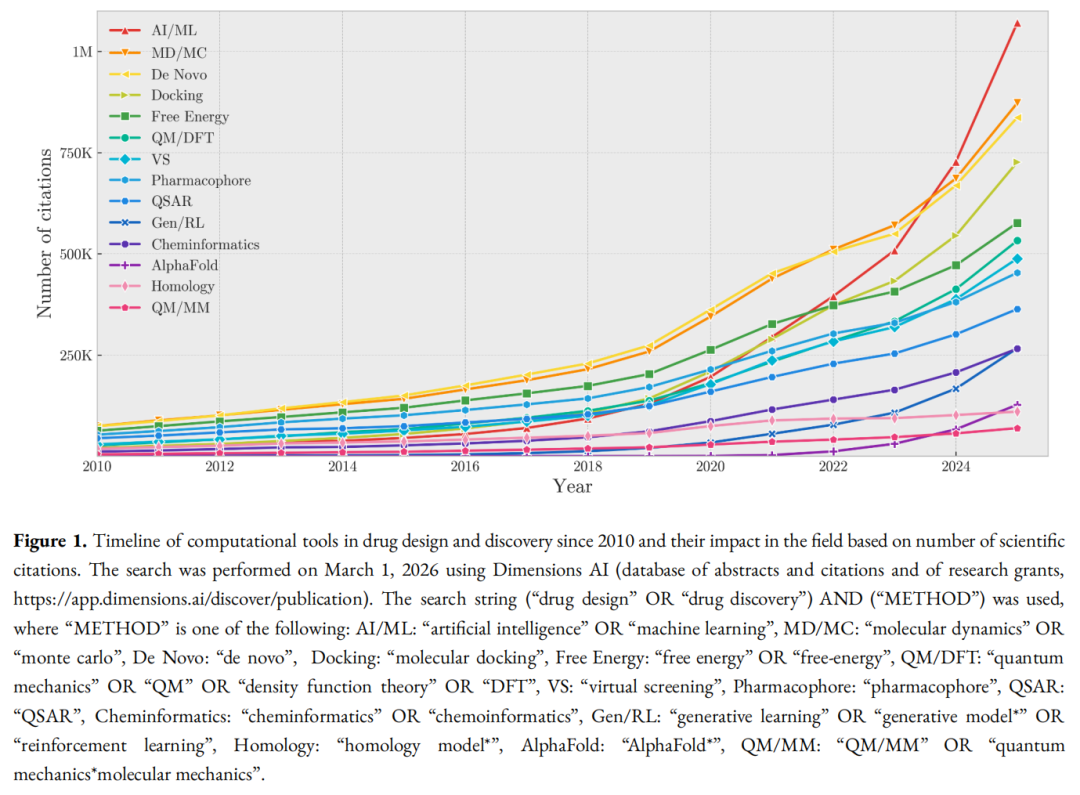
关键洞察:历史上没有任何一种方法单独"革命"了药物发现,进步总是来自多种方法的协同整合。当前 AI 浪潮须在这个连续演进的视角下被审视,而非被视为范式断裂。
1.3 药物发现数据的特殊挑战
论文明确指出,将 AI 方法从 NLP/CV 迁移到药物发现时面临以下根本性挑战:
- • 数据规模悬殊:药物发现数据集与 LLM 训练语料相比,体量差距达数个数量级
- • 数据质量问题:公开数据集中错误和不一致性普遍存在;专有数据集的生成成本极高
- • 自监督学习受限:与语言模型不同,药物发现数据代表的是物理世界的客观事实,难以借助 RLHF 等人类反馈机制规模化扩张
- • 靶点特异性:结合亲和力等关键数据高度依赖具体靶点,难以跨靶点泛化
- • 分布外泛化:最有价值的预测往往是对前所未有的靶点或化学空间的推断,而这正是 ML 模型最脆弱的地方
作者援引 Sutton 的"苦涩的教训"(Bitter Lesson):通用的、可随算力规模化扩展的学习方法,长期来看总是优于领域专用的定制方案。但由于上述约束,药物发现领域距离真正享受这一红利可能还需要相当时间。
二、论文框架与方法论
2.1 "大挑战"框架的历史传统
论文有意识地将自身定位于"大挑战"(Grand Challenge)的学术传统中——从希尔伯特的数学问题(1900),到 CASP 蛋白质结构预测竞赛,到 SAMPL、D3R、CACHE 等药物发现专项竞赛。这类框架的核心价值在于:将模糊愿景转化为可共同检验的具体问题,进而汇聚跨学科资源。
2.2 统一的挑战描述模板
论文为每项挑战采用统一的六要素框架,这本身就是一种方法论贡献:
挑战定义 → 精确的输入/输出规格(相当于一道工程题的题面)
药物发现关联 → 解决该挑战能影响哪个决策节点、降低哪类风险
关键问题 → 当前方法的技术瓶颈所在
历史与现状 → 方法演进的脉络与当前能力边界
输入/输出规格 → 标准化的数据格式要求
评估指标 → 可量化的进展标准(而非"更好了"的定性判断)2.3 范围界定
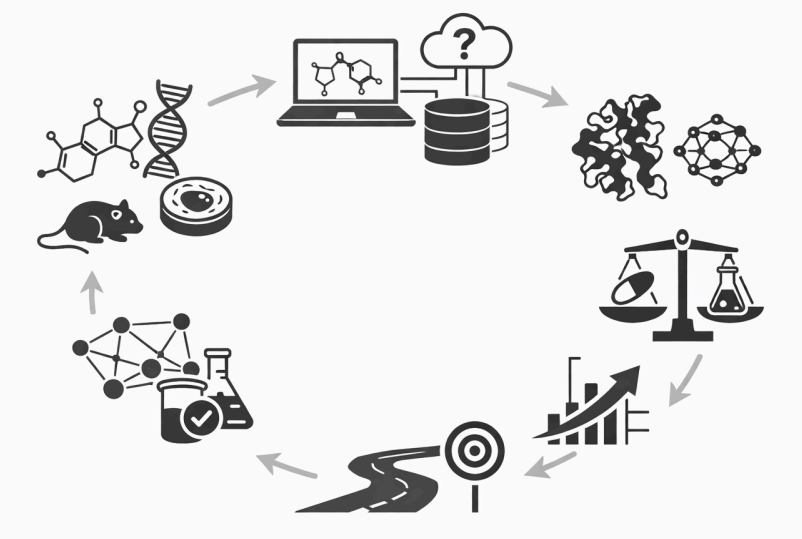
论文聚焦于小分子药物发现的 Discovery 阶段,即从靶点确定到 IND 候选物提名,覆盖:靶点选择 → 苗头化合物发现 → 苗头到先导 → 先导优化 → IND 前研究。不涉及临床试验、商业化及生物制剂等模态。
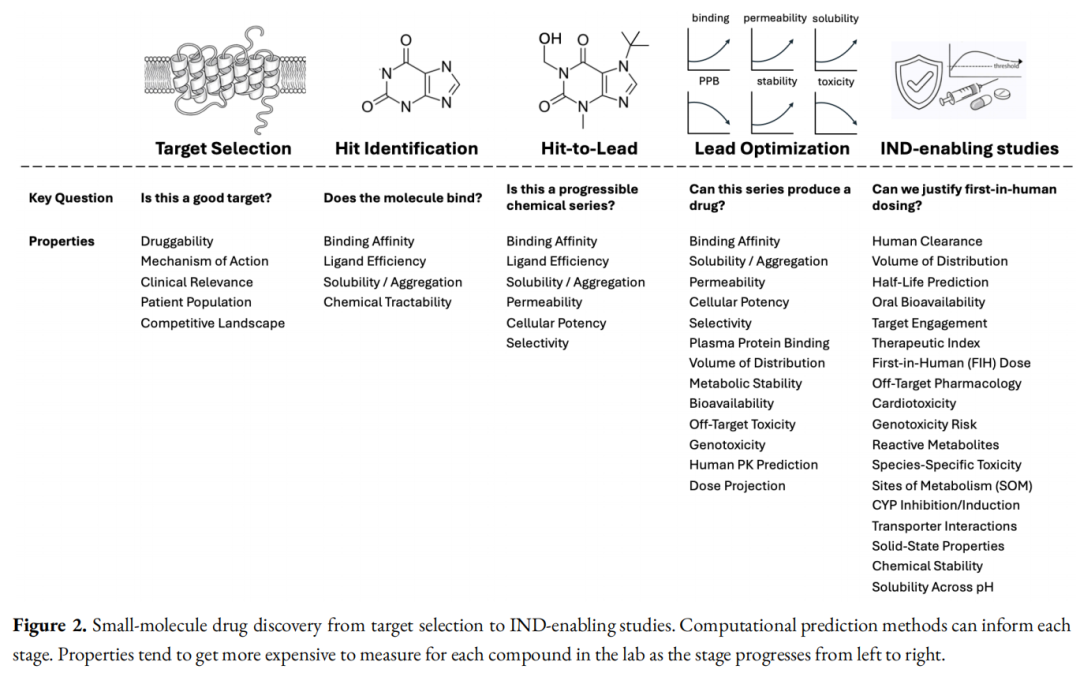
三、化学领域:从分子设计到实物合成
3.1 合成路线规划(发现化学)
核心问题:给定一个小分子结构,预测其合成路径与实验操作规程,达到目标纯度(筛选阶段 ≥90%,先导优化 ≥95%,IND ≥99%)。
药物发现相关性:在 DMTA(设计-合成-测试-分析)循环中,"Make"步骤往往是最耗时、最耗资源的环节。计算辅助合成规划的价值在于将虚拟设计与物理实现的成本降低,同时也是按需虚拟化合物库(make-on-demand library)的可行性前提。
当前瓶颈:
- • 逆合成分析本质上是搜索问题,但化学无"确定性规则",产物预测(包括产率和副产物)的准确性仍是核心难点
- • 现有数据驱动工具严重依赖专利数据库,而这些数据的分子复杂度远低于现代药物化学需求
- • 常用 benchmark(如 USPTO-50k)的准确率已接近平台期,但并不代表真实能力的提升——"分数好看"≠"化学家会用"
- • 多步合成、立体化学控制、天然产物类骨架的规划仍是公认难题
- • 工具输出停留在"建议路线"层面,缺乏完整的操作程序(试剂用量、温度、时间、纯化方案等)
评估指标:反应产物/条件/产率预测的定量准确率;盲测化学家偏好研究(多步合成);预测路线是否能在实验室实际走通(终极指标,但不具可扩展性)。
3.2 合成路线规划(工艺化学)
核心问题:在发现化学合成规划的基础上,额外考虑可扩展性、可持续性、成本效益和纯度控制,以满足临床试验及商业化生产需求。
与发现化学的差异:工艺化学不只是"多合一点",还涉及安全性(避免危险中间体)、Green Chemistry 指标(PMI、E-Factor)、设备适配性、精制和结晶方案等完全不同的优化目标。
现状评估:计算机辅助工艺化学的研究基础极为薄弱。机器学习用于路线优化尚处于探索阶段,与石化行业成熟的流程模拟工具(如 Aspen Plus)相比,制药工艺设计工具差距显著。
3.3 共价结合
核心问题:给定靶蛋白,预测具有选择性共价抑制剂的化学结构;并预测其结合亲和力、反应动力学(kinact)及脱靶效应。
重要性:共价药物在肿瘤、免疫和感染领域已有大量获批案例(如奥希替尼、伊布替尼)。其优势在于持续靶点占据(sustained target engagement),特别适合需要持续抑制的生物学场景。
技术挑战:
- • 弹头(warhead)电亲核性的精确调控:反应性太强→脱靶毒性;太弱→无效
- • 亲核位点微环境建模:同一类反应性残基(如半胱氨酸)在不同蛋白口袋中的反应活性差异可达多个数量级
- • 竞争反应预测:谷胱甘肽等细胞内亲核体的消耗会降低药效或引发毒性
- • QM/MM 方法精度与计算成本的权衡:全量子力学处理对实际尺寸的蛋白体系目前仍不可行
3.4 化学稳定性与降解产物
核心问题:给定药物分子或制剂,预测在特定环境条件下的降解动力学、降解途径和降解产物。
关键性:稳定性是药物的关键质量属性(CQA),直接影响药效、安全性和货架期。目前监管机构不允许完全依赖计算预测替代长期稳定性实验(通常需要 12 个月),但计算工具可以指导结构修饰和配方优化,显著缩短实验周期。
挑战要点:固态与溶液态降解的差异、辅料相容性的预测、光降解和氧化降解动力学的建模均缺乏成熟的通用工具。
四、结构领域:从序列到三维空间
4.1 配体晶型与多晶型
核心问题:给定化学结构,预测所有热力学稳定的晶体堆积形式(多晶型、水合物、溶剂化物及盐型),并基于自由能对其稳定性进行排序。
药物发现相关性:多晶型直接影响溶解度、溶出速率和生物利用度。历史上最著名的案例是 Ritonavir(1998 年):一种新的、溶解度更低的多晶型在商业化后突然出现,导致药物下架——损失逾 2.5 亿美元,彻底改变了制药界对固态筛选的重视程度。
评估指标体系:
- • 结构精度:RMSD₃₀(30 分子超胞的均方根偏差),阈值通常 <0.5 Å
- • 晶胞参数匹配:单元格尺寸、角度和空间群的准确率
- • 热力学排序:全局最小晶型识别率(GMIR),即实验观测的最稳定晶型是否被预测为能量最低结构
- • 多晶型全集回收率(PRR):所有已知晶型是否均被预测枚举覆盖
竞赛基准:Cambridge Crystallographic Data Centre(CCDC)主办的 CSP(Crystal Structure Prediction Blind Test)是该领域最权威的国际竞赛,目前顶尖方法在中等大小刚性分子上表现良好,但对柔性分子和盐型仍具挑战。
4.2 蛋白质结构预测
核心问题:给定氨基酸序列,预测蛋白质的三维结构或构象系综。
AlphaFold 时代的重新定位:AlphaFold2(2021)和 RoseTTAFold 的出现从根本上重塑了该领域的问题边界。对于序列与训练集中已知结构同源的蛋白,预测精度已接近实验方法。但以下子问题仍是公认难点:
子问题 | 当前状态 | 主要挑战 |
|---|---|---|
膜蛋白 | 较弱 | 脂质双层环境难以建模 |
本征无序蛋白(IDP) | 较弱 | 无固定结构,系综描述需求 |
大型复合体(>MDa) | 中等 | 多链装配精度 |
配体诱导的构象变化 | 差 | 训练以 apo 结构为主 |
蛋白质-配体共折叠 | 新兴 | 下一节重点讨论 |
标准评估指标(CASP 竞赛体系):
- • TM-score(模板建模评分):>0.5 认为整体折叠正确;>0.9 接近实验精度
- • GDT-TS(全局距离测试总评分):基于多阈值残基匹配比例的综合评分
- • LDDT(局部距离差异测试):无需叠合的局部原子环境精度评估,对侧链堆积和局部特征更敏感
4.3 蛋白质构象动力学与自由能面
核心问题:给定蛋白质三维结构,预测在指定能量阈值内的所有可访问构象及其相对自由能。
为何重要:蛋白质是动态的,静态结构只是一个快照。构象动力学控制着:隐蔽口袋的暴露与闭合、变构信号的传递、结合诱导的构象选择。忽视蛋白质动力学是早期计算药物发现的重大局限之一。
主要方法及局限:
方法 | 优势 | 局限 |
|---|---|---|
常规 MD | 真实物理模型 | 时间尺度受限(~μs),难以逾越高能垒 |
增强采样(Metadynamics, AMD) | 加速慢运动采样 | 需要先验知识定义集体变量 |
粗粒化模型 | 可达 ms 尺度 | 牺牲原子精度 |
AI 方法(如 MSM, Boltzmann Generator) | 速度快 | 大多仍处于概念验证阶段 |
实验对照手段:NMR 弛豫、氢氘交换质谱(HDX-MS)、室温 X 射线晶体学、Cryo-EM 粒子系综重权重——这些实验方法本身的通量和标准化程度也制约着计算方法的基准建立。
4.4 蛋白-配体复合物结构预测
核心问题:给定蛋白质序列和小分子化学结构,预测结合复合物的三维坐标(结合姿态,binding pose)。
重要性:正确的结合姿态是基于结构的药物设计(SBDD)的基础——如果对接姿态错误,基于结构的设计、虚拟筛选打分以及下游的自由能计算都将建立在错误的前提上。
方法演进:
- • 传统分子对接(Glide、AutoDock、GOLD 等):速度快,适合大规模虚拟筛选;但对柔性配体、诱导契合效应和极性口袋处理不足
- • 诱导契合对接(IFD):允许蛋白侧链和局部骨架响应配体,准确性显著提升,但计算成本高
- • 共折叠方法(AlphaFold3、Chai-1、Boltz-1 等):将蛋白-配体对接纳入端到端结构预测,2023 年后快速发展,在某些基准上超过传统对接
现存根本局限:
- • 训练分布外的新型骨架(novel chemotypes)泛化能力仍差
- • 正确的结合姿态≠正确的结合亲和力(见下章能量领域)
- • PLINDER、CASF、CELPP 等基准集已经暴露出评估体系本身存在数据泄露问题
4.5 隐蔽口袋识别
核心问题:给定蛋白质 apo 态(无配体)结构,预测在 holo 态(有配体)结构中出现的、在 apo 态中被掩盖或不存在的可成药结合位点。
战略意义:隐蔽口袋极大地扩展了"可成药蛋白质组"(druggable proteome)。KRAS 长期被视为"不可成药靶点",正是 KRAS-G12C 的隐蔽口袋(Switch II pocket)被发现后,才催生了索托拉西布(Sotorasib)等突破性药物。
核心技术挑战:
- • 口袋形成是罕见的动力学事件,在正常 MD 时间尺度上难以捕获
- • 需要同时准确建模蛋白质呼吸运动和界面溶剂化/去溶剂化效应
- • 隐蔽口袋的形状依赖于具体配体,存在"鸡和蛋"问题
评估指标:空间重叠评分(Dice/Jaccard 系数)、口袋中心距离、检测率/精确率/召回率/F1 分数;以及在给定构象系综中的口袋形成概率预测。
五、能量领域:分子识别的热力学与动力学
5.1 蛋白-配体结合亲和力与排序
核心问题:给定蛋白质三维结构和一系列配体,预测其结合自由能(ΔG,单位 kcal/mol)或相对结合自由能(ΔΔG),实现准确的活性排序。
为什么这是最重要的挑战之一:结合亲和力直接决定所需剂量,进而影响安全窗口和治疗指数。虚拟筛选的核心目标就是在合成/购买化合物之前进行活性预排序,减少无效的实验消耗。
方法精度梯度:
方法类别 | 代表方法 | 精度(ΔΔG RMSE) | 速度(相对) | 适用场景 |
|---|---|---|---|---|
经验打分函数 | Glide SP/XP | ~2–3 kcal/mol | 极快 | 大规模虚拟筛选 |
MM-GBSA/PBSA | 再打分 | ~1.5–2 kcal/mol | 中等 | 小规模重评估 |
自由能微扰(FEP) | FEP+, RBFE | ~1 kcal/mol | 慢 | 先导优化系列 |
热力学积分(TI) | OpenFE | ~1 kcal/mol | 慢 | 同上 |
机器学习打分 | NNScore, ΔΔG-NET | 依赖数据质量 | 快 | 有足够数据时 |
根本局限:现有力场不包含金属离子相互作用、极化效应(polarization)和质子转移;FEP 的计算成本限制了其在分子骨架跳跃(scaffold hop)中的应用;ML 模型的"哑铃效应"(dumbbell effect)——训练集亲和力范围过宽——会人为夸大评估指标。
5.2 蛋白-配体结合选择性
核心问题:预测小分子在靶蛋白与脱靶蛋白(off-targets)之间的结合选择性谱图,尤其是可靠地区分"强结合"与"可忽略结合"。
重要性:选择性差是药物毒性和临床失败的主要驱动因素之一。对于激酶类靶点,人类基因组编码约 500 种激酶,高度同源的激酶口袋对选择性设计构成极大挑战。
问题的本质难点:
- • 挑战不仅是计算 ΔΔG,更是在能量差异很小(往往仅 1–2 kcal/mol)时可靠区分结合与不结合
- • 蛋白质构象的完整性:许多脱靶蛋白没有高质量晶体结构,AlphaFold 结构的适用性仍在评估中
- • 规模问题:一个化合物需要对蛋白质组中数百至数千个潜在靶点进行评估
- • 实验选择性谱图本身不完整:任何实验面板只覆盖有限靶点,未知脱靶永远存在
评估框架:ROC-AUC(全域判别性能)、富集因子(EF,前 n% 富集率)、MCC(马修斯相关系数,处理样本不平衡)、RMSE/MAE(定量亲和力差)。
5.3 蛋白-配体结合动力学
核心问题:给定已知结合的靶蛋白和配体,预测结合速率常数(kon)和解离速率常数(koff),进而推算驻留时间(residence time = 1/koff)。
为何驻留时间有时比亲和力更重要:在受体脱敏(receptor resensitization)慢于解离速率的系统中,koff 比 Kd 更能预测体内药效。大量临床案例表明,具有相似 Kd 的化合物因 koff 不同而显示出截然不同的持续时长和安全性。
计算挑战:配体解离(unbinding)过程往往发生在微秒至毫秒乃至秒的时间尺度,远超传统 MD 的可达范围。增强采样方法(τ-RAMD、WeTMD、Metadynamics)在特定系统上有效,但通用性和校准精度仍是问题。
5.4 变构调控与激动/拮抗效应
核心问题:给定蛋白质 apo 结构,预测配体与远端(非正交)位点结合如何通过变构机制调制蛋白功能,并预测该配体是激动剂、拮抗剂还是部分/偏向激动剂。
治疗意义:变构位点通常在蛋白家族间保守性低,因此靶向变构位点有望获得更高选择性。变构调节已在 GPCRs、激酶、蛋白酶、核受体等多类靶点中得到验证。
科学难点:变构信号传递涉及远距离构象耦合(allosteric communication pathway),这要求模型能够捕获蛋白质内的集体运动和热力学耦合——对于 MD 和 AI 方法都是极高难度要求。目前已发表的变构位点预测验证案例大多是回顾性的(retrospective),前瞻性验证极为稀缺。
六、药理学领域:从分子到体内行为
药理学领域集中了最多的挑战(约 10 项),也是最贴近临床转化决策的部分。每项挑战的"准确预测"都能直接降低昂贵的体内实验消耗。
6.1 pKa 预测
核心问题:预测分子在水溶液及蛋白结合位点中的 pKa 值和质子化状态分布。
为何是所有 ADME 预测的前提:溶解度、脂溶性(logD)、渗透性、血浆蛋白结合乃至结合亲和力,都依赖于正确的质子化状态。pKa 预测出错将引发下游所有预测的系统性偏差——"垃圾进,垃圾出"在这里体现得最为直接。
技术挑战:
- • 互变异构体的枚举和优先级排序
- • 含多个可电离基团的分子(多质子分子)的耦合电离
- • 含硫、卤素等杂原子的杂环结构预测精度偏低
- • 溶解度极低化合物的实验 pKa 测量误差本身较大,影响模型训练
当前精度(SAMPL6 等竞赛数据):
- • 顶尖方法 RMSE:~0.7–1.0 pKa 单位(对应约 5–10 倍平衡常数误差)
- • 多数方法 RMSE:1–3 pKa 单位
- • 注:1 个 pKa 单位的误差在药物化学决策中通常可以接受,但对精确建模(如自由能计算输入)而言误差仍然显著
6.2 聚集、溶解度与渗透性
核心问题:预测分子在两相系统(水溶液 vs 脂质膜)中的分布行为,包括水溶性(logS)、胶体聚集倾向和被动膜渗透性(Caco-2 Papp)。
三属性的相互依赖与权衡:
- • 溶解度↑通常意味着 logP↓,而 logP↓通常伴随渗透性↓
- • 聚集体可以"消耗"游离药物浓度,造成虚假的低渗透性和体外非特异性蛋白结合
- • 溶解度预测需要同时考虑固态(晶体能)和液态(溶剂化自由能)性质——而前者在早期研发中通常未知
方法精度(外部测试集):
- • logS(水溶性):ML 方法 R² ≈ 0.7–0.8,RMSE ≈ 0.6–0.8 log 单位
- • 渗透性(Caco-2):分类准确率约 75–85%
- • 聚集预测:基于 MD 的方法近期报告97% 分类准确率(明显优于化学信息学过滤器的75%)
新兴难点:超出"五规则"(bRo5)范围的大环肽、PROTACs 等新模态分子的溶解度/渗透性预测,以及非平衡态的过饱和/沉淀动力学建模。
6.3 血浆蛋白结合(PPB)与分布容积(Vd)
核心问题:预测化合物的血浆蛋白结合率(%PPB 或 fup)和表观分布容积(Vd)。
临床意义:只有游离(unbound)药物才能与靶点结合、被代谢和被排泄。fup 是连接体外测定和体内药代动力学的关键参数。Vd 决定了单次给药后组织中的暴露程度,对体半衰期和给药频率设计至关重要。
预测困难点:
- • 极端结合(fup <1% 或 >90%)的预测精度尤差,而这两类恰好临床上最重要
- • 非特异性吸附(对实验设备的 NSB)在高亲脂性化合物中严重干扰实验数据质量
- • Vd 预测依赖于组织-血浆分配系数(Kp)的准确获得,而 Kp 本身难以实验直接测量
- • 物种差异:大鼠、小鼠、犬、猴到人的 PPB 种差显著,跨种外推错误会放大剂量预测误差
评估标准:Vd 预测的"2 倍误差内"(within 2-fold)通常被认为是可接受水平,反映了该问题的固有不确定性量级。
6.4 代谢稳定性/清除率
核心问题:预测化合物的代谢稳定性,量化为内在清除率(CLint)、肝脏清除率(CLhep)或代谢半衰期(t1/2)。
核心地位:代谢稳定性是口服生物利用度的重要决定因素之一(通过首过效应),也是给药频率设计和毒性风险评估的基础参数。
多层次挑战:
分子层面 → 酶底物识别(CYP 家族的广谱底物耐受性)
酶动力学层面 → K_m, k_cat 的准确预测
体外系统层面 → 微粒体蛋白非特异性结合(NSB 校正)
体内外相关性(IVIVE) → 从微粒体→肝脏→整体的多级缩放
整体药代层面 → 肝脏外清除(肾、肠、肺等)的贡献当前 ML 模型表现:分类准确率约 75–85%(稳定 vs 不稳定),但对 CLint 的定量预测精度仍较低;对超出训练集化学空间的新骨架泛化能力是公认软肋。
6.5 口服生物利用度
核心问题:给定小分子结构,预测口服给药后到达体循环的药物分数(F%)。
为何口服生物利用度是"最难预测的单一参数":F 是溶解度、渗透性、首过肝脏代谢、肠道代谢、转运体外排等多个独立过程的乘积——任何一个环节的预测误差都会被放大。
其中 是肠道吸收分数, 是肠道代谢后剩余分数, 是肝脏提取率。
数据质量问题:
- • 实验数据具有严重的选择偏倚——性质差的化合物往往在进行口服给药实验之前就被淘汰
- • 数据分布极端偏斜(许多化合物 F<10% 或 F>80%),回归建模极其困难
混合策略的前景:ML 预测 ADME 各子参数(溶解度、渗透性、清除率)→ 输入 PBPK 模型进行机制性整合,比直接端到端预测 F 的方法更具可解释性和物理基础。
6.6 药物代谢:代谢位点与代谢物预测
核心问题:预测药物分子在特定物种中被代谢酶修饰的具体位点(SOM),以及生成的代谢物结构。
临床意义:代谢"软点"(metabolic soft spots)的识别指导结构修饰(如氘代、引入位阻)以提高稳定性;活性代谢物可能具有独立疗效或毒性;反应性代谢物(reactive metabolites)是特异质肝毒性的重要机制。
技术复杂性:
- • CYP 家族(尤其是 CYP3A4、2D6、2C9)具有极宽的底物谱,同一化合物可被多个 CYP 亚型并行代谢
- • I 相代谢(氧化、还原、水解)和 II 相代谢(葡萄糖醛酸化、硫酸化)的机制截然不同,缺乏统一建模框架
- • 代谢物验证需要 LC-MS 或 NMR,实验通量较低,标注数据稀缺
评估指标:Top-N 准确率(实验验证的 SOM 是否出现在预测排名的前 N 位);代谢物结构的精确/亚结构匹配;CYP 亚型归属准确率。
6.7 毒性预测
核心问题:给定小分子结构,预测其在多种毒性机制维度上的风险,包括但不限于 hERG 心脏毒性、遗传毒性/致癌性、肝毒性(DILI)、肾毒性、反应性代谢物等。
毒性不是单一问题:这是一个由多个机制异质性极高的子挑战构成的集合,每个子挑战都需要独立建立基准。常见的毒性机制分类:
毒性类型 | 主要机制 | 预测难度 |
|---|---|---|
hERG 心脏毒性 | 钾通道阻断 → QT 延长 | 中等(有大量数据) |
遗传毒性 | DNA 损伤、Ames 测试 | 中等 |
肝毒性(DILI) | 代谢激活、线粒体损伤、免疫反应 | 高(特异质性 DILI 极难预测) |
肾毒性 | 多种机制 | 高(数据少) |
发育毒性 | 复杂 | 高 |
近期监管动向:FDA 于 2024 年宣布计划逐步淘汰单克隆抗体的动物测试强制要求,进一步推动体外和计算毒性预测的发展,但小分子的相关政策尚未跟进。
特别强调:假阴性(漏报真实毒性)比假阳性的后果严重得多,因此该领域的模型校准(calibration)和特异性/灵敏度的权衡至关重要。
6.8 剂量预测
核心问题:基于临床前体外数据和体内 PK/PD 数据,预测人体安全有效的给药方案(起始剂量、剂量范围和给药间隔)。
战略重要性:首次人体(FIH)剂量的选择直接影响 I 期临床的效率和安全性。基于 PBPK 模型的机制性剂量预测已成为监管申报(IND)的标准支持内容。
多层次挑战:
化合物层面:PK 参数的精确预测(CL, Vd, t1/2, F)
靶点层面:靶组织中的药物浓度(target engagement)
物种外推层面:临床前→人的跨物种缩放(allometric scaling 的局限性)
患者层面:个体间变异(遗传多态性、年龄、疾病状态)
特殊人群:儿科、老年、肝肾损伤患者的剂量调整PBPK 模型的现状:已在监管机构(FDA、EMA)获得广泛认可。但模型的预测质量高度依赖输入参数(尤其是 fup、CLint)的质量——"垃圾进,垃圾出"定律在此同样适用。标准验收标准:关键 PK 参数预测值在实验值 2–3 倍以内。
6.9 药代动力学/药效动力学(PK/PD)建模
核心问题:整合 PK 参数(药物浓度-时间曲线)和 PD 参数(靶点结合、生物标志物响应、疗效指标),预测给定给药方案下的时间-效应关系,并优化剂量方案。
作为整合框架的价值:PK/PD 建模是将"分子如何进入人体"(PK)和"分子如何改变人体"(PD)两个知识体系定量连接的桥梁。它是:
- • 早期候选化合物评估的理性框架
- • 转化医学(临床前→临床)的核心工具
- • 监管申报(MIDD,Model-Informed Drug Development)的重要支柱
新兴方向:量化系统药理学(QSP)将 PK/PD 框架扩展至信号通路、疾病进展和联合用药建模;AI 驱动的虚拟患者(virtual patient populations)模拟患者间变异;个性化给药算法整合基因组生物标志物。
七、横切主题与系统性反思
7.1 数据质量是一切的前提
贯穿全文的一个核心认识是:不是方法不够先进,而是数据不够好。具体体现在:
- • 公开数据集中的系统性错误(测量误差、单位错误、混淆变量)
- • 实验条件不标准化导致跨实验室/跨数据库的可重复性差
- • 选择偏倚:被收录进数据库的化合物本身并非随机样本
- • 专有数据集的壁垒:最高质量的数据掌握在少数大型制药公司手中
7.2 决策驱动而非精度驱动
论文一再强调,计算方法的价值不在于"精度高"本身,而在于改变了项目决策。这意味着:
- • 一个在 benchmark 上 R²=0.85 的模型,如果其误差分布恰好使最坏的化合物看起来最好,则毫无价值
- • 富集率(enrichment factor)、决策阈值处的分类精度,往往比全数据集上的相关系数更能反映实际价值
- • "适配用途"(fit-for-purpose)的评估框架优于通用精度评估
7.3 物理方法与数据驱动方法的互补
论文没有站队"物理方法 vs AI",而是清醒地指出:
- • 物理方法(FEP、MD、QM/MM):机理可解释,具有外推能力,但计算成本高,通量受限
- • 数据驱动方法(ML、DL):速度快,可规模化,但分布外泛化能力弱,缺乏物理约束
- • 最有前景的路径是物理约束的机器学习(physics-informed ML):将物理先验注入 ML 框架,兼顾速度与可靠性
7.4 基准建设的重要性与现有缺陷
现有基准(benchmarks)存在系统性问题:
- • 训练集与测试集时间泄露(temporal leakage):评估时使用了训练时间之后才发表的结构
- • 化学空间同质性:许多竞赛数据集集中在相似的化学类型,低估了真实多样性
- • 回顾性 ≠ 前瞻性:大多数评估是回顾性的,而真实价值在于前瞻性预测
论文呼吁建立更多盲测(prospective blinded)的前瞻性评估竞赛,如 CASP 之于蛋白质结构预测的地位。
八、对不同受众的启示
8.1 计算化学家与 AI for Drug Discovery 研究者
- • 优先选择具有清晰决策意义的问题,而非仅仅刷新 benchmark 成绩
- • 投入资源建立高质量的领域专用数据集,而非仅仅改进模型架构
- • 主动参与前瞻性竞赛(SAMPL、CACHE、CELPP 等),用真实预测能力说话
- • 深化对"误差如何影响下游决策"的理解——1 kcal/mol 的误差在不同场景中影响截然不同
8.2 药物化学家与项目团队
- • 理解计算工具的真实能力边界,避免对黑盒预测过度依赖或过度排斥
- • 配体结合亲和力的 FEP 预测已达相当精度,但对骨架跳跃和新型化学类型仍需谨慎解读
- • 代谢稳定性和毒性预测的分类精度(~80%)意味着:可以有效过滤最差候选物,但不能替代真实测量
- • pKa 预测的误差会系统性地传播至几乎所有下游 ADME 预测,错误的质子化状态必须尽早纠正
8.3 生物技术创业公司与投资人
论文提供了一个评估"AI 药物发现"技术主张的参照系:
主张 | 参照本文结论 | 成熟度判断 |
|---|---|---|
"AI 可以预测结合亲和力" | FEP 已相当成熟;纯 ML 对新骨架仍差 | 需区分问题边界 |
"AI 可以设计合成路线" | 对简单路线可用;多步复杂路线仍差 | 中等成熟 |
"AI 可以预测毒性" | 高通量分类有效;特异质毒性几乎无法预测 | 端点依赖性强 |
"AI 可以预测口服生物利用度" | 分类可行;定量预测精度仍低 | 早期工具 |
"AlphaFold 解决了结构问题" | apo 态静态结构很好;动力学/配体诱导构象/共折叠仍是开放问题 | 部分解决 |
8.4 监管机构与政策制定者
- • 清晰定义的挑战和量化指标有助于制定模型辅助药物开发(MIDD)的接受标准
- • 鼓励建立行业共享的高质量基准数据集(类似 OpenTargets 在靶点领域的作用)
- • 逐步推进替代动物实验(New Approach Methodologies, NAMs)需要同步提高计算毒理学的标准化程度
九、总结评价
9.1 本文的学术贡献
这篇论文的贡献不在于提出新方法,而在于提供了一个经过严格审视的、可供社区共同操作的框架。具体贡献包括:
- 1. 问题目录化:将药物发现中多个分散的、定义模糊的计算问题,统一为具有标准结构的挑战描述
- 2. 指标规范化:为每项挑战提出可量化的评估标准,使"进步"有迹可查
- 3. 能力边界厘清:基于一线实践者的访谈,诚实评估了当前方法的真实局限,而非文献综述中常见的过度乐观
- 4. 多方向激励:为计算化学家、AI 研究者、实验科学家和资金提供者提供了协调一致的方向性指引
9.2 局限性
- • 聚焦于小分子药物发现,不涵盖抗体、多肽、PROTACs、RNA 靶向药物等新兴模态
- • 多数挑战的目标指标是研究者设定的,尚需通过未来竞赛和实践来校验是否适切
- • 部分领域(如 QSP、组织特异性毒性)的挑战定义相对笼统,仍需进一步细化
9.3 最终判断
在 AI 制药的喧嚣中,这篇文章选择了做一件更难、更有价值的事:把问题说清楚。它不是宣言,也不是路线图,而更像一份集体认知的诚实盘点——我们到底在哪里,还差多远,用什么方法量。
这种稀缺的清醒,加上参与者横跨学术、工业和风险投资的广泛代表性,使本文成为计算药物发现领域近年来最具参考价值的元文献之一。无论你是从事这个领域的研究者,还是试图理解 AI 药物发现真实进展的观察者,都值得认真阅读。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-25,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号