circlize系列(六):复现Science单细胞互作弦图添加受配体表达量(基于cellchat分析结果)

circlize系列(六):复现Science单细胞互作弦图添加受配体表达量(基于cellchat分析结果)

KS科研分享与服务-TS的美梦
发布于 2026-04-02 13:07:08
发布于 2026-04-02 13:07:08
论文图:
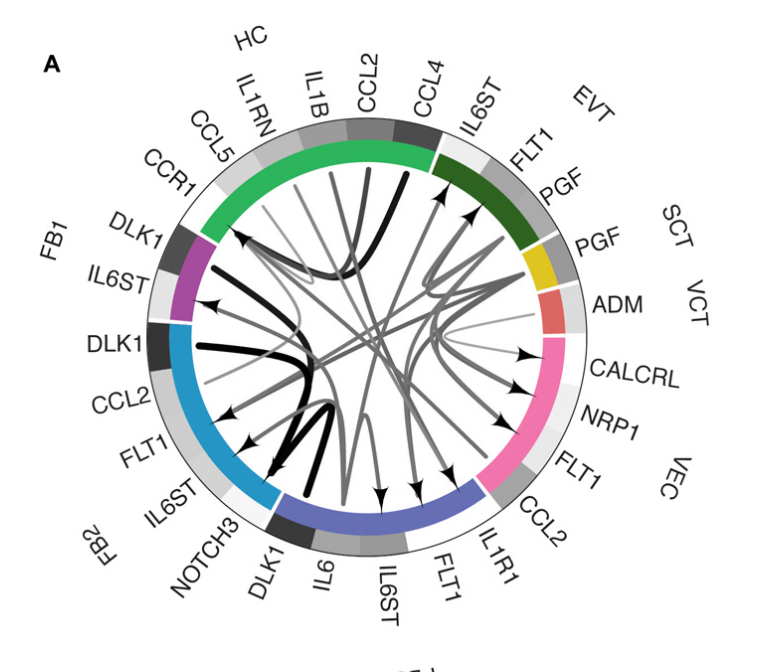
复现图:
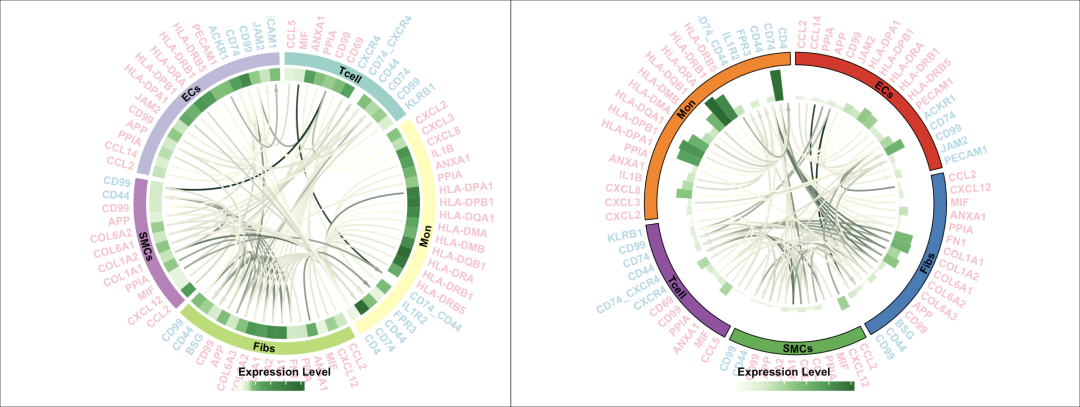
circlize作图比较麻烦的地方在于代码很长,如果切换数据修改的内容比较繁琐。所以将这个过程包装为一个函数,方便使用,当然更个性化的展示需要自己往里面添加参数和设置。演示使用的是cellchat分析得到的结果,输入数据是cellchat object。
1、提取数据
可以自定义提取需要展示的受配体,使用cellchat的subsetCommunication函数,提取需要关注的细胞互作结果,并筛选目标展示,比如这里我们演示就设置了source和target cells,并且只展示互作强度大于0.01的数据。
library(CellChat)
HD.cellchat <- readRDS("~/Downloads/HD.cellchat.rds")
unique(HD.cellchat@idents)
# [1] Kers Mon Tcell lang Men Fibs SMCs ECs Mast
# Levels: ECs Fibs Kers lang Mast Men Mon SMCs Tcell
HD.com <- subsetCommunication(HD.cellchat, sources.use = c("Tcell","Mon","Fibs","SMCs","ECs"),
targets.use = c("Tcell","Mon","Fibs","SMCs","ECs"))
#为了演示顺利不繁琐,我们对prob做了筛选,实际按照自己的想法即可,这里仅仅是为了减少结果
HD.com <- HD.com[HD.com$prob > 0.01,]
HD.com <- HD.com[,1:5]2、整理画图数据
library(dplyr)
library(tidyr)
result_df <- HD.com %>%
pivot_longer(
cols = c(ligand, receptor),
names_to = "group",
values_to = "gene"
) %>%
mutate(
cells = if_else(group == "ligand", source, target)
) %>%
select(gene, group, cells) %>%
distinct() # 去重
result_df <- result_df %>%
mutate(
cells = factor(cells, levels = c("Tcell","Mon","Fibs","SMCs","ECs")),
group = factor(group, levels = c("ligand",'receptor'))
) %>%
arrange(cells, group) # 先按celltype排序,再按group排序#设置celltype颜色
group_colors <- c("Tcell" = "#0e9c23",
"Mon" = "#6aada3",
"Fibs" = "#b03c64",
"SMCs" = "#bccf42",
"ECs" = "#e08214")
result_df$color <- group_colors[result_df$cells]
#设置受配体颜色
LR_color <- c("ligand" = "pink",
"receptor" = 'lightblue')
result_df$LR_color <- LR_color[result_df$group]3、画图
#====================================================================================
# 1、初始化作图
#====================================================================================
circos.clear()#清空当前作图,便于新的circle plot
group_size <- table(result_df$cells)#这个是每个细胞大群也就是分组的size,这里就是包含的亚群数目,需要注意这个涉及到后面扇形分区,所以顺序要对
#设置布局
circos.par(start.degree = 90, cell.padding = c(0, 0, 0, 0), #起始位置,扇区内行距为0
gap.after = 2,#设置每个扇区之间的gap,前面的扇区之间小一点,最后两个扇区也就是首尾的位置扇区开头大一点
circle.margin = c(0.1, 0.1, 0.1, 0.1))#环形图距离画布的距离
#初始化plot
circos.initialize(factors = result_df$cells,#扇区scctor,这是已经排好序的数据
xlim = cbind(0, group_size))#每个扇区xlim,每个扇区元素不同,所以每个扇区的xlim是0到扇区元素长度
#====================================================================================
#2、添加第一轨道,受配体名称及区分
#====================================================================================
#plot最外层受配体基因,并区别颜色
circos.track(
ylim = c(0, 1), #y轴范围
bg.border = NA, #不要背景
track.height = 0.01,#贵高高度
panel.fun = function(x, y) {
sector_index = get.cell.meta.data("sector.index") #获取当前扇区index
group_size = group_size[sector_index] #获取当前扇区长度
#适用循环plot 文字,因为是多个扇区
for (i in 1:group_size) {
circos.text(
x = i - 0.5, #位于中间
y = 0.5, #y轴位置
labels = result_df$gene[result_df$cells == sector_index][i], #标注,索引到扇区对应的亚群
col= result_df$LR_color[result_df$cells == sector_index][i],
font = 2,#文字加粗
facing = "reverse.clockwise", #文字排布方式向外
niceFacing = TRUE,
adj = c(1, 0.5),
cex = 0.8)#文字大小
}
}
)
#====================================================================================
#3、添加第二轨道,受配体celltype注释
#====================================================================================
#第二轨道,添加celltype注释
##因为我们是按照扇区来plot的,所以添加group注释就很简单了
circos.track(ylim = c(0, 1),
bg.border = NA,
track.height = 0.08,
bg.col=group_colors,#分组注释背景颜色
panel.fun=function(x, y) {
xlim = get.cell.meta.data("xlim") #获取当前扇区x,y范围
ylim = get.cell.meta.data("ylim")
sector.index = get.cell.meta.data("sector.index")#扇区索引
circos.text(mean(xlim),#取mean,文字位于中心
mean(ylim),
sector.index, #label就是扇区索引
col = "black", #文字颜色
cex = 0.8,
font=2,
facing = 'bending.inside',
niceFacing = TRUE)
})
#====================================================================================
#3、添加第三轨道,受配体表达量
#====================================================================================
#这里使用cellchat object直接提取表达量
# 提取需要展示受配体基因的表达量(平均表达)
all_genes <- unique(result_df$gene) # 获取所有需要展示的基因
# 使用CellChat的computeAveExpr函数提取平均表达量
expr_matrix <- computeAveExpr(HD.cellchat,
features = all_genes,
type = "truncatedMean",
trim = 0.1)
# 转换为数据框格式,便于后续使用
expr_df <- as.data.frame(expr_matrix)
expr_df$gene <- rownames(expr_df)
# 将长格式转换为宽格式(基因×细胞类型)
expr_long <- expr_df %>%
pivot_longer(cols = -gene,
names_to = "cells",
values_to = "expression")
# 匹配result_df中的顺序
result_df <- result_df %>%
left_join(expr_long, by = c("gene", "cells"))
# 设置表达量的颜色映射函数
# 使用从浅到深的渐变颜色
col_expr_fun <- colorRamp2(
c(0,
quantile(result_df$expression, 0.25, na.rm = TRUE),
quantile(result_df$expression, 0.5, na.rm = TRUE),
quantile(result_df$expression, 0.75, na.rm = TRUE),
max(result_df$expression, na.rm = TRUE)),
c("#f7fcf0", "#c7e9c0", "#74c476", "#31a354", "#006d2c") # 绿色渐变,从浅到深
)
# 添加表达量热图轨道
circos.track(
ylim = c(0, 1),
bg.border = NA,
track.height = 0.08, # 轨道高度
panel.fun = function(x, y) {
sector_index = get.cell.meta.data("sector.index") # 获取当前扇区
group_data = result_df[result_df$cells == sector_index, ] # 获取当前扇区的数据
n_genes = nrow(group_data) # 当前扇区的基因数量
if(n_genes > 0) {
# 为每个基因绘制色块
for(i in 1:n_genes) {
# 获取表达量值
expr_value <- group_data$expression[i]
# 绘制色块(整个矩形区域填充颜色)
circos.rect(
xleft = i - 1, # 左边界
xright = i, # 右边界
ybottom = 0, # 底部
ytop = 1, # 顶部(填满整个高度)
col = col_expr_fun(expr_value), # 颜色映射
border = NA # 无边框,更整洁
)
}
}
}
)
#====================================================================================
#3、最后添加互作连线
#====================================================================================
##添加互作连线
HD.com <- HD.com %>%
mutate(
source = factor(source, levels = c("Tcell","Mon","Fibs","SMCs","ECs"))
)%>%
arrange(source)
col_fun = colorRamp2(range(HD.com$prob), c("#FFFDE7", "#013220"))
# 添加互作连线
for(i in 1:nrow(HD.com)) {
# 获取起始位置信息
source <- as.character(HD.com$source[i]) # 确保转换为字符
ligand <- as.character(HD.com$ligand[i])
# 找到在 result_df 中的索引(确保因子水平一致)
from_subset <- result_df[result_df$cells == source, ]
from_idx <- which(from_subset$gene == ligand)
# 获取目标位置信息
target <- as.character(HD.com$target[i])
receptor <- as.character(HD.com$receptor[i])
to_subset <- result_df[result_df$cells == target, ]
to_idx <- which(to_subset$gene == receptor)
if(identical(ligand, receptor)==FALSE){
# 计算在各自扇区中的位置(从0开始计数)
from_pos <- from_idx - 0.5
to_pos <- to_idx - 0.5
}else{
from_pos <- from_idx[1] - 0.5
to_pos <- to_idx[2] - 0.5
}
# 添加连线,这里需要注意,其实有些函数里面,箭头plot出来不是很好,肯定相干参数调整
circos.link(
sector.index1 = source, # 起始扇区
point1 = from_pos, # 起始位置
sector.index2 = target, # 目标扇区
point2 = to_pos, # 目标位置
col = col_fun(HD.com$prob[i]), # 连线颜色,互作强度
lwd = 2,#粗细
directional = 1,#连线箭头,0表示没有箭头,1表示从point1 to point2方向箭头,-1则相反。2表示双向箭头
arr.length=0.2,#箭头长度
arr.width=0.1#箭头宽度
)
}
很显然很长,如果换个数据又要替换很多内容,光代码就很繁琐。所以自定义函数:这个函数还有很多可以自定义的地方,比如颜色,形状等。不过使用它也可以直接、轻松完成比较好的绘图。
函数输入需要是cellchat object,以及筛选好的可视化文件,都是基于cellchat。至于图的展示提供了两种方式,热图+柱状图二选一!完整内容已发布微信VIP,请自行下载!

测试一下:假如这是筛选好的数据。
HD.com <- subsetCommunication(HD.cellchat, sources.use = c("Tcell","Mon","Fibs","SMCs","ECs"),
targets.use = c("Tcell","Mon","Fibs","SMCs","ECs"))
HD.com <- HD.com[HD.com$prob > 0.01,]
HD.com <- HD.com[,1:5]自定义顺序,celltype颜色,以及表达量展示方式:
ks_cellchat_LRplot(cellchat_obj = HD.cellchat,
select_LR = HD.com,
exp_shpe ='heatmap',
celltype_order = c("Tcell","Mon","Fibs","SMCs","ECs"),
group_colors = c("#8DD3C7", "#FFFFB3","#B3DE69","#BC80BD","#BEBADA"))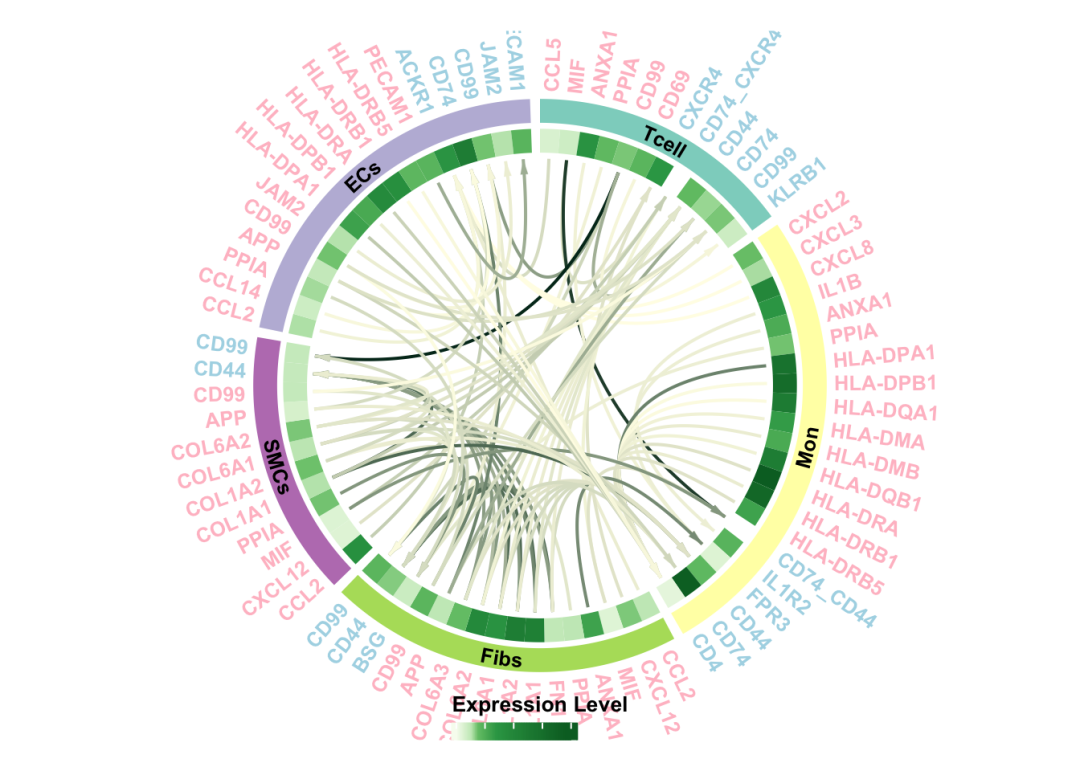
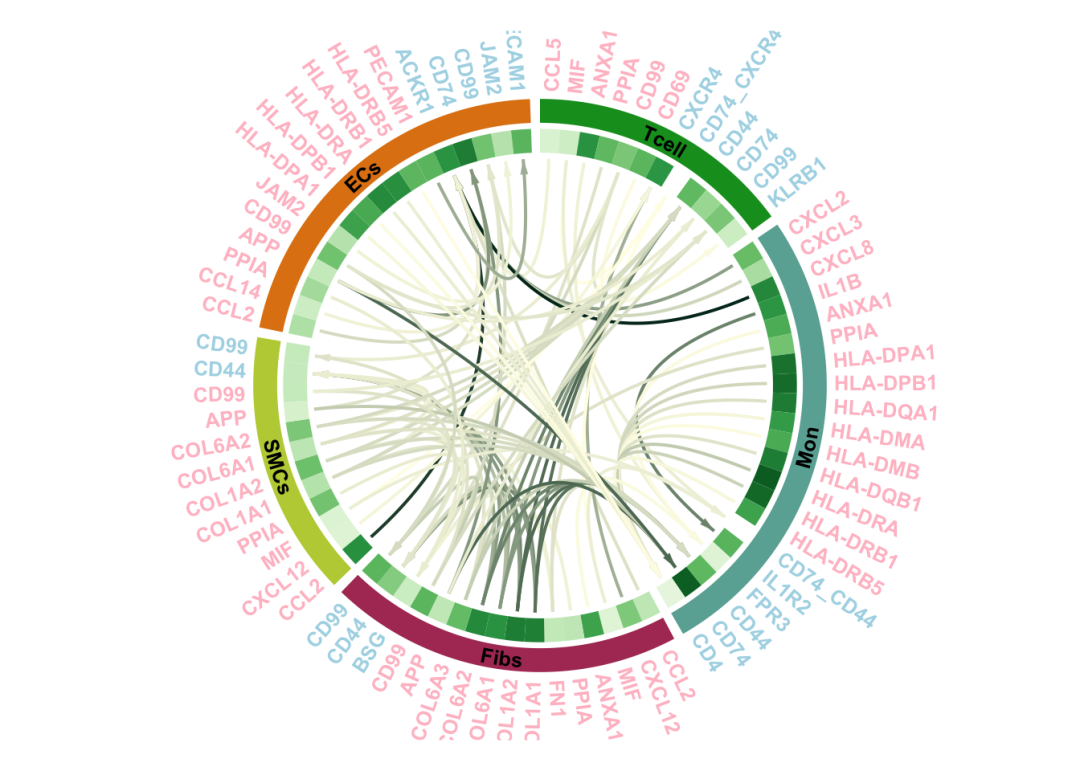
颜色,顺序按照默认结果,这里表达量使用柱状图展示:
ks_cellchat_LRplot(cellchat_obj = HD.cellchat,
select_LR = HD.com,
exp_shpe ='barplot',
bg.border='black')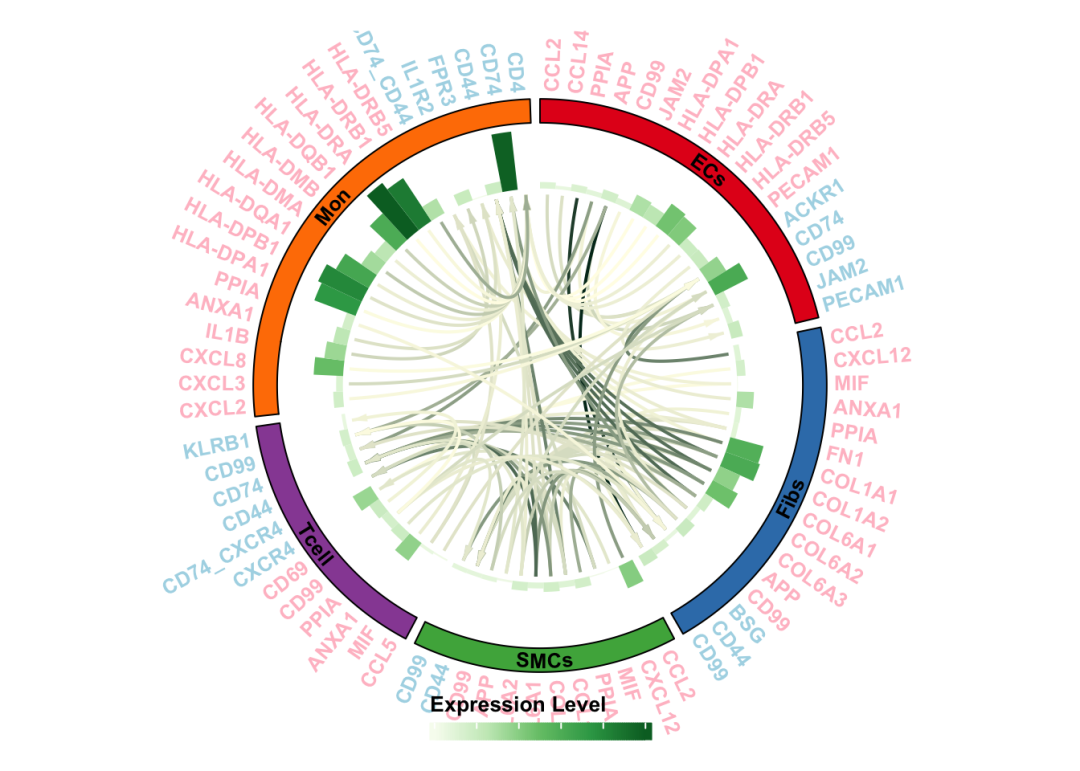
结果完美,函数画起来方便多了!唯一的问题就是legend需要手动调整!
觉得分享有用的点个赞再走呗!
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-22,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号