Nat. Commun. | 当图神经网络学会说 我不确定 ——不确定性量化驱动的高效分子设计

Nat. Commun. | 当图神经网络学会说 我不确定 ——不确定性量化驱动的高效分子设计

DrugIntel
发布于 2026-04-13 15:12:54
发布于 2026-04-13 15:12:54

推荐论文: Lung-Yi Chen & Yi-Pei Li. Uncertainty quantification with graph neural networks for efficient molecular design. Nature Communications, 2025, 16: 3262. DOI:10.1038/s41467-025-58503-0 代码:github/Lung-Yi/uncmoo
一、引言:为什么这篇论文值得关注?
计算辅助分子设计(Computational-Aided Molecular Design, CAMD)正在从"经验驱动"走向"数据驱动"。图神经网络(GNN)因其能直接在分子图上学习结构-性质关系而成为该领域的核心工具。然而,当我们用训练好的 GNN 作为代理模型(surrogate model),并借助优化算法在广阔的化学空间中搜索最优分子时,一个根本性的矛盾浮现了:
优化过程天然倾向于将搜索推向训练数据稀疏的未知区域——恰恰是模型预测最不可靠的地方。
模型在这些区域可能给出过于自信却严重偏离真实值的预测,导致优化方向被误导。这不是个别现象,而是所有基于代理模型的分子优化框架共同面临的系统性挑战,学术上称之为域外迁移(domain shift)问题。
本研究系统地将不确定性量化(Uncertainty Quantification, UQ)集成到基于有向消息传递神经网络(D-MPNN)和遗传算法(GA)的分子优化框架中,并在 19 个分子设计基准任务上进行了全面评估。其核心发现是:概率改进优化(Probabilistic Improvement Optimization, PIO)策略在大多数任务中显著优于不考虑不确定性的传统方法和另一种 UQ 集成策略(期望改进,EI),尤其在多目标优化中优势明显。
二、研究背景:CAMD 的技术演进与瓶颈
2.1 分子设计的优化范式
现代 CAMD 将分子设计建模为优化问题:分子结构是变量,目标性质是优化目标。主流方法包括:
- • 生成模型路线: 变分自编码器(VAE)将分子编码到连续潜空间进行采样与优化;基于 SMILES 的循环神经网络(RNN)结合强化学习进行目标导向生成。这些方法依赖预训练,在训练数据有限或分布狭窄时,生成分子的多样性可能受限。
- • 直接优化路线: 遗传算法(GA)直接在分子图或 SMILES/SELFIES 字符串上进行变异和交叉操作,无需预训练生成模型。GA 天然维持种群多样性,对小数据集友好,计算开销相对较低,适合直接探索化学空间。
本文选择后者——GA + GNN 代理模型的组合,绕开了生成模型的训练瓶颈,直接在开放化学空间中搜索。
2.2 代理模型的信任边界问题
GNN(特别是 D-MPNN)作为代理模型已展现出强大的分子性质预测能力。然而,所有数据驱动模型都面临一个根本局限:在训练分布之外的预测不可靠。
传统的贝叶斯优化(BO)框架通过高斯过程回归(GPR)自然地提供不确定性估计,但 GPR 的计算复杂度为 ,难以扩展到大规模数据集。虽然存在稀疏近似、随机特征展开等加速策略,但在需要处理大规模化学空间的场景下,参数化模型(如 GNN)因其固定参数量而具有天然的可扩展性优势。
然而,**将 UQ 集成到参数化模型中用于开放化学空间的优化——而非仅用于虚拟筛选或主动学习——是否有效?如何实现最优集成?**这正是本文试图回答的核心问题。
2.3 相关工作的空白
此前的研究已将 UQ 与参数化模型结合用于主动学习和虚拟筛选,提升了工作流效率。但有研究指出,在定义良好的化合物库中进行虚拟筛选时,不考虑不确定性的贪心策略有时表现出与 UQ 方法相当甚至更优的性能。这是否意味着 UQ 在分子设计中并不必要?
本文的关键区别在于:它考察的不是有限化合物库中的虚拟筛选,而是开放化学空间中的持续优化——搜索轨迹不断更新,模型面临的外推程度远超虚拟筛选场景。在这种条件下,UQ 的价值需要被重新审视。
三、方法论:三位一体的技术框架
3.1 代理模型:D-MPNN
本文采用 Chemprop 中实现的有向消息传递神经网络(Directed Message Passing Neural Network, D-MPNN)作为代理模型。D-MPNN 的核心设计包括:
- • 有向消息传递机制: 在分子图的有向边上进行消息传递,根据分子连接性更新隐藏原子状态,避免了无向消息传递中信息回流(tottering)的问题。
- • 分子指纹生成: 对所有隐藏原子向量求和或取平均,生成固定维度的分子指纹。
- • 前馈神经网络: 以分子指纹为输入,预测目标性质。
与依赖固定分子描述符的传统模型不同,D-MPNN 直接从分子图中学习结构表示,能捕捉原子间详细的连接和空间关系。
3.2 不确定性量化:两种方法
论文评估了 Chemprop 中实现的两种 UQ 方法,均能将总不确定性分解为偶然不确定性(aleatoric)和认识不确定性(epistemic):
方法一:深度集成 + 均值-方差估计(Ensemble + MVE)
- • 认识不确定性(): 训练 个不同初始化种子的模型,计算预测间的方差:
- • 偶然不确定性(): 每个模型额外输出一个神经元预测数据依赖的不确定性,训练时使用高斯分布的负对数似然(NLL)作为损失函数:
集成模型中各模型的偶然不确定性取平均。
方法二:Evidential Learning
- • 无需训练多个模型,通过单模型直接预测 Normal Inverse-Gamma(NIG)分布的四个参数 。
- • 偶然不确定性:
- • 认识不确定性:
两种方法的总不确定性均为 。
实际选择策略: 论文发现两种方法在不同数据集上各有优劣——MVE 在反应数据集上容易发散,Evidential Learning 在有机发光体数据集上精度下降。最终对有机发光体任务使用 Ensemble + MVE,其余任务使用 Evidential Learning。
3.3 适应度函数设计:核心创新
这是论文的技术核心。作者系统比较了三种单目标适应度函数和四种多目标适应度函数。
单目标适应度函数
(1)直接目标最大化(DOM)——基线方法
其中 是代理模型对分子 的预测均值,(最大化)或 (最小化)。这是传统的贪心策略,完全不考虑不确定性。
(2)概率改进优化(PIO)——本文推荐方法
PIO 计算的是分子 的性质值超过预设阈值 的概率,输出值在 之间。它天然融合了预测均值和不确定性两方面的信息。
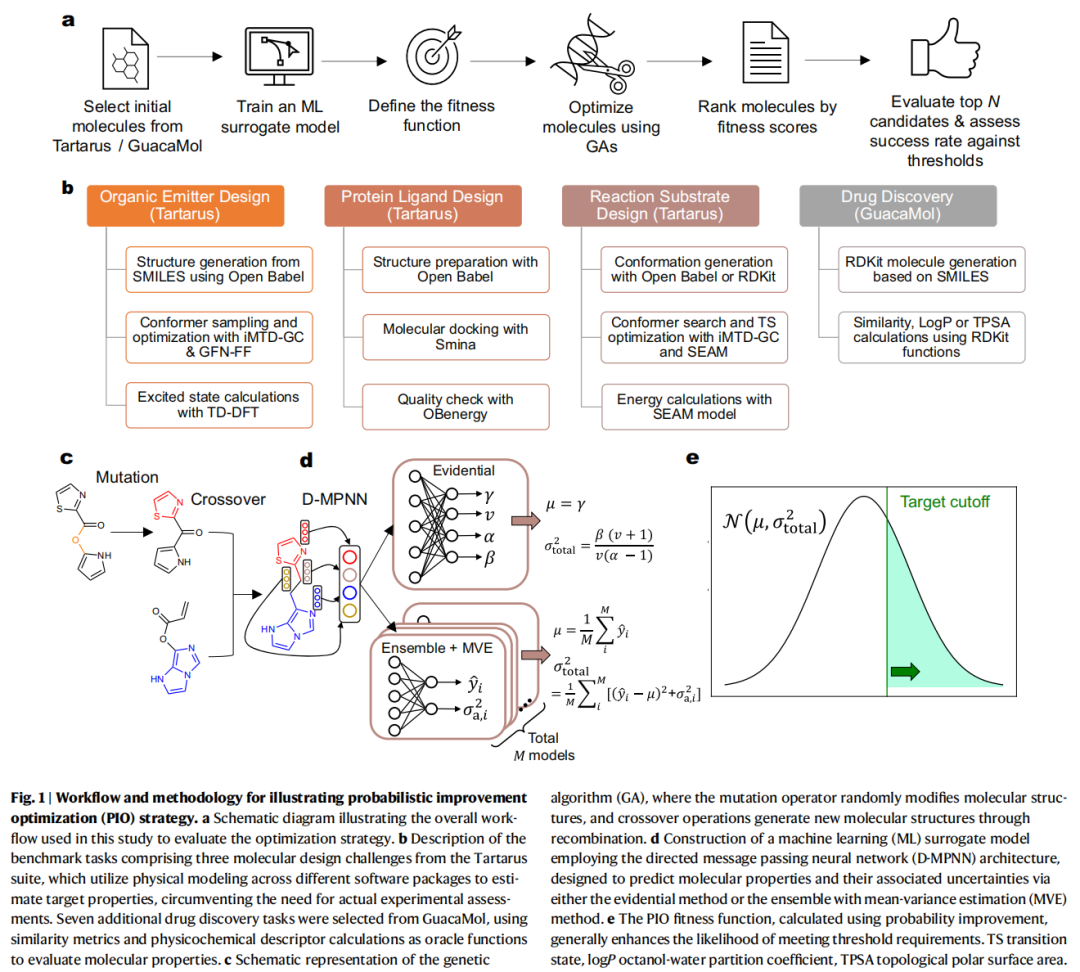
(3)期望改进(EI)
EI 不仅考虑超过阈值的概率,还加权了超过的幅度。它是贝叶斯优化中最经典的采集函数。
多目标适应度函数
(1)加权求和(WS)
以各目标在数据集中分布标准差的倒数为权重。
(2)归一化曼哈顿距离(NMD)
将超出阈值的目标视为等同优秀,只惩罚未达标的部分。
(3)NMD-WS 混合方法
当所有目标未达标时使用 NMD,达标后切换为 WS 继续优化。
(4)PIO 多目标扩展——本文核心贡献
将各目标的达标概率相乘。这一设计的精妙之处在于:任一目标的达标概率接近零,整体适应度就趋近于零,天然实现了目标间的平衡约束,避免了加权方法中某些目标被过度牺牲的问题。
3.4 遗传算法:Janus
分子优化采用 Janus 算法,基于 SELFIES 分子表示进行操作。相比 SMILES,SELFIES 的关键优势在于:任何文本修改都能保持化学合法性,即使随机变异也不会产生无效分子结构。
实验设置为:以 Top-10,000 分子为初始种群,进行 15 次独立优化运行,每次 10 个迭代,每迭代引入 500 个新分子。最终合并所有运行结果以降低 GA 固有的随机性影响。
四、实验评估
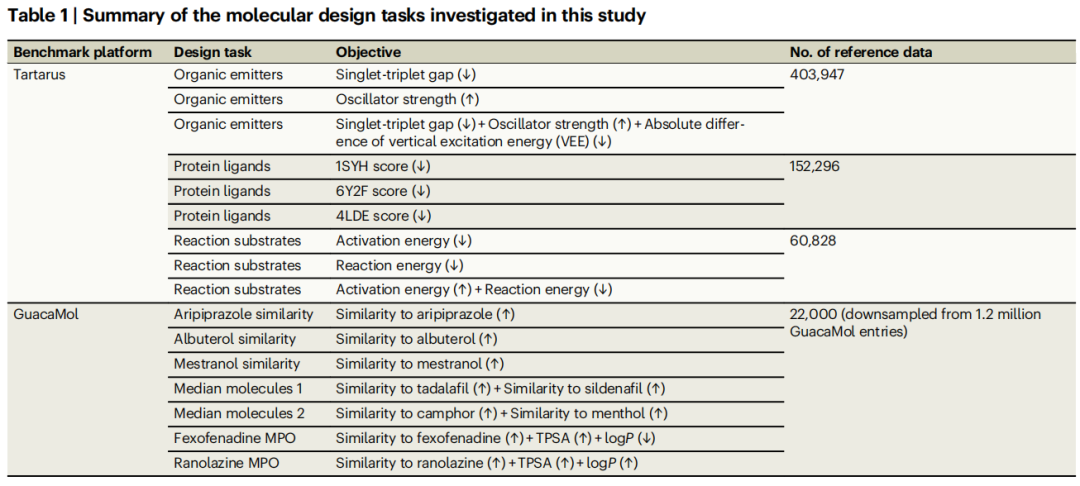
4.1 基准平台与任务
论文在两个权威基准平台上进行了系统评估,覆盖 19 个数据集、16 个优化任务:
Tartarus 平台(物理模拟验证)
设计类别 | 任务 | 计算方法 | 数据规模 |
|---|---|---|---|
有机发光体 | 单重态-三重态间隙(↓)、振子强度(↑)、三目标联合优化 | 构象采样 + 半经验QM + TD-DFT | 403,947 |
蛋白质配体 | 1SYH / 6Y2F / 4LDE 对接得分(↓) | 分子对接 + 经验打分函数 | 152,296 |
反应底物 | 活化能(↓)、反应能(↓)、双目标联合优化 | 力场优化 + SEAM过渡态方法 | 60,828 |
Tartarus 的核心特点是使用真实的物理模拟(力场、DFT、分子对接)作为验证函数,包含随机构象搜索和对接采样带来的数据噪声,更贴近真实研究场景。
GuacaMol 平台(药物发现)
设计类别 | 任务 | 验证方法 | 数据规模 |
|---|---|---|---|
药物相似性 | 阿立哌唑 / 沙丁胺醇 / 麦雌醇相似性(↑) | RDKit Tanimoto 相似度 | 22,000(降采样) |
中位分子 | 他达拉非+西地那非 / 樟脑+薄荷醇 中位相似性(↑) | RDKit 相似度 | 同上 |
多性质优化 | 非索非那定 MPO / 雷诺嗪 MPO | 相似度 + logP + TPSA | 同上 |
GuacaMol 使用确定性的 RDKit 函数计算性质值,无数据随机性。论文有意将数据集降采样至 22,000 以模拟代理模型不完美的实际场景。
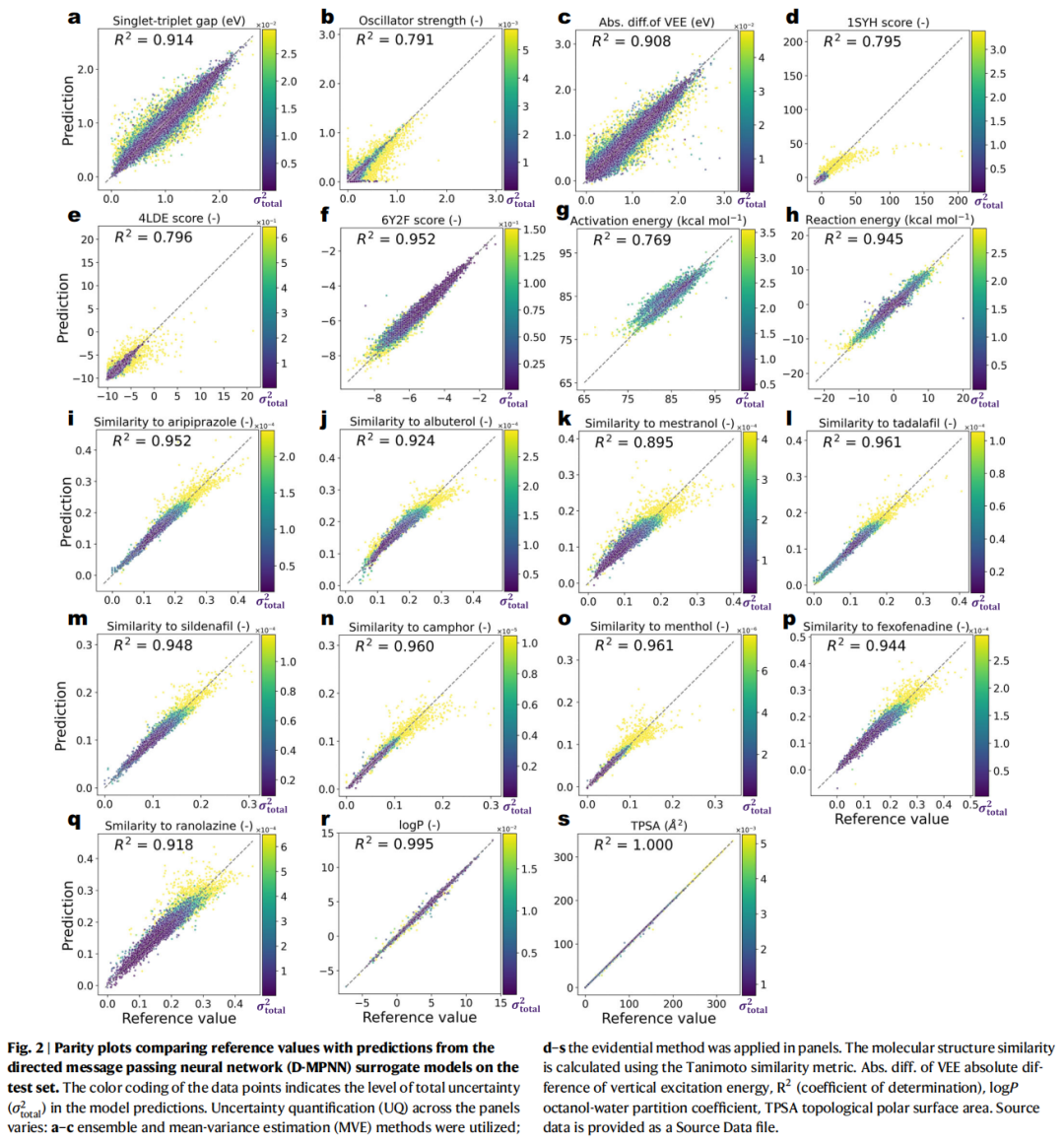
4.2 代理模型性能验证
在投入优化实验之前,论文首先验证了 D-MPNN 的预测能力和 UQ 校准质量:
- • 预测精度: 所有任务的测试集 R² 值在 0.769–1.000 之间,D-MPNN 能有效捕捉性质趋势(Fig. 2)。
- • UQ 校准: 基于置信度的校准曲线(Fig. 3)显示,所有测试集的校准误差曲线下面积(AUCE)均低于 0.1,表明残差分布与高斯假设基本吻合,不确定性估计在测试集上是可靠的。
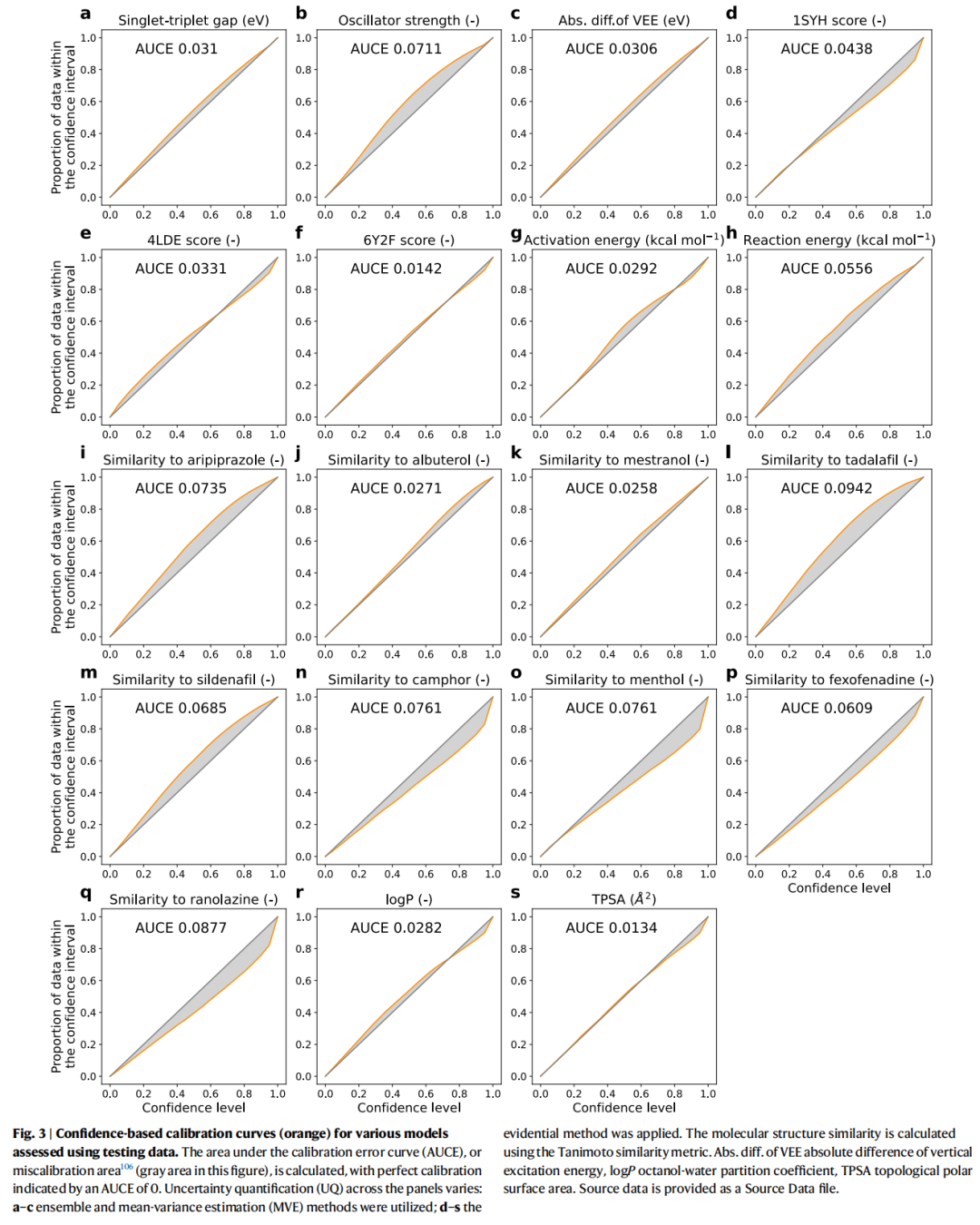
这一步骤至关重要——它建立了 PIO 方法的基础假设(预测服从已知均值和方差的高斯分布)在实践中的合理性。
4.3 单目标优化结果
以下是关键任务的 Top-K 命中率对比(命中率 = 真实性质超过阈值的分子比例):
任务 | DOM (Top-10) | EI (Top-10) | PIO (Top-10) | DOM (Top-100) | EI (Top-100) | PIO (Top-100) |
|---|---|---|---|---|---|---|
振子强度(↑) | 0.20 | 0 | 0.30 | 0.16 | 0.12 | 0.21 |
4LDE 得分(↓) | 0.40 | 0 | 0.90 | 0.49 | 0.03 | 0.92 |
6Y2F 得分(↓) | 0.40 | 0 | 0.80 | 0.50 | 0 | 0.49 |
反应能(↓) | 0.50 | 0.70 | 0.90 | 0.40 | 0.57 | 0.76 |
阿立哌唑相似性(↑) | 0.50 | 0 | 1.00 | 0.53 | 0.06 | 0.58 |
沙丁胺醇相似性(↑) | 0 | 0 | 1.00 | 0.03 | 0 | 0.62 |
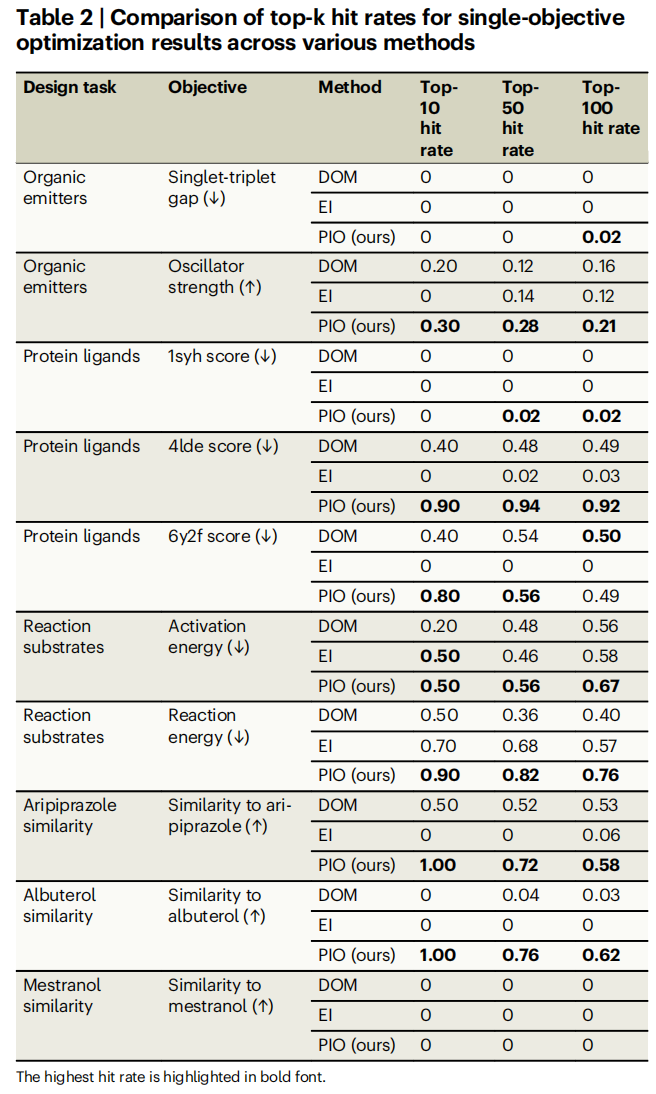
核心发现:
- • PIO 在绝大多数任务中取得最高命中率。 尤其在阿立哌唑和沙丁胺醇相似性任务中,PIO 的 Top-10 命中率达到 100%,而 DOM 和 EI 表现平平。
- • EI 在大多数任务中表现不佳,甚至不如不考虑不确定性的 DOM。 这是一个重要的反直觉发现。
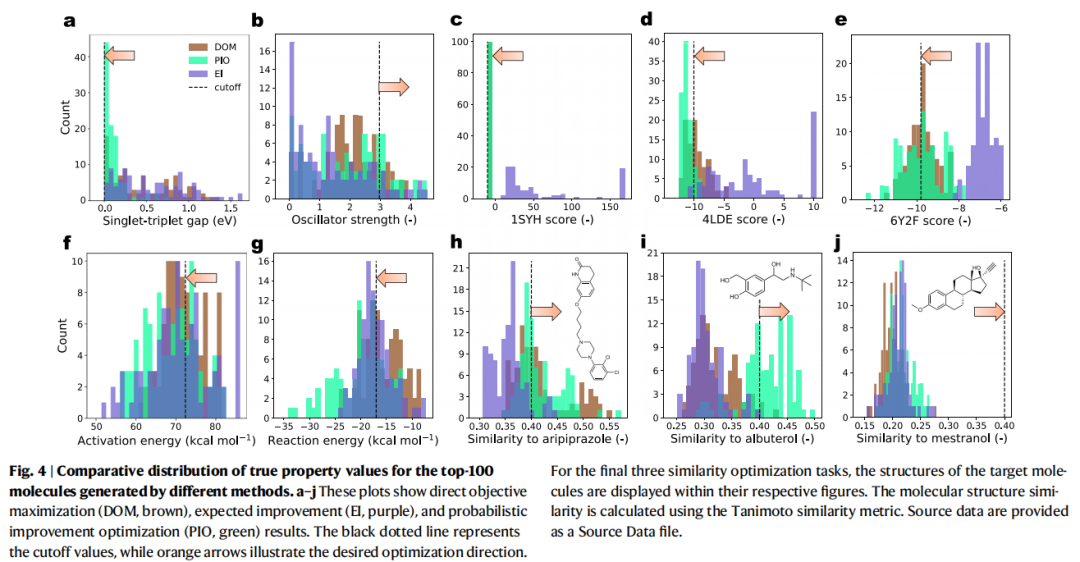
4.4 为什么 EI 失灵而 PIO 胜出?
论文通过 parity plot 分析(Fig. 5)揭示了机制层面的原因:
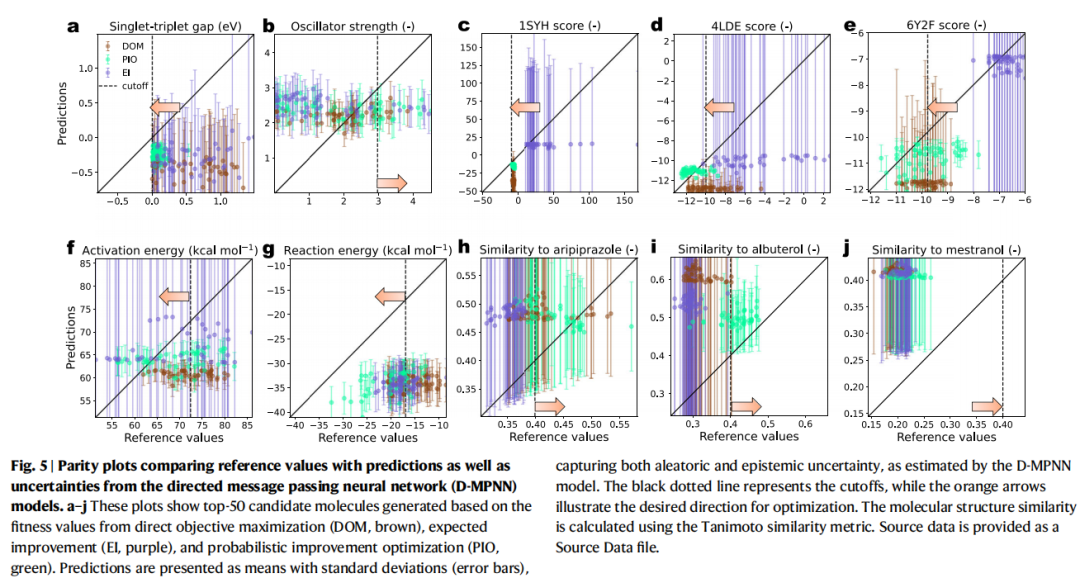
- • DOM 选出的分子倾向于具有极端的预测均值(最高或最低),被推向外推区域,预测偏差大。
- • EI 选出的分子倾向于具有最高的不确定性。因为 EI 的公式中,当预测均值相近时,高不确定性的分子会获得更高的期望改进值。在化学空间极大的场景下,D-MPNN 会给大量候选结构分配高不确定性,导致 EI 的"期望改进"被严重膨胀,引导搜索偏向高方差区域。
- • PIO 选出的分子在预测均值合理的同时保持较低的不确定性。PIO 的输出值天然在 之间有界,不会被极端方差放大,使优化过程更加稳定可靠。
这一发现对整个领域具有重要启示:EI 在传统 BO(小搜索空间、少量迭代、GPR 模型)中的成功经验,不能直接迁移到大规模化学空间中的 GNN 优化场景。
4.5 多目标优化结果
任务 | WS (Top-10) | NMD (Top-10) | NMD-WS (Top-10) | PIO (Top-10) |
|---|---|---|---|---|
有机发光体三目标 | 0 | 0.60 | 0 | 0.80 |
反应底物双目标 | 0.20 | 0.10 | 0.40 | 0.40 |
中位分子 1 | 0 | 0.60 | 0.90 | 0.90 |
非索非那定 MPO | 0 | 0.40 | 0.10 | 0.30 |
雷诺嗪 MPO | 0 | 0 | 0 | 0.20 |
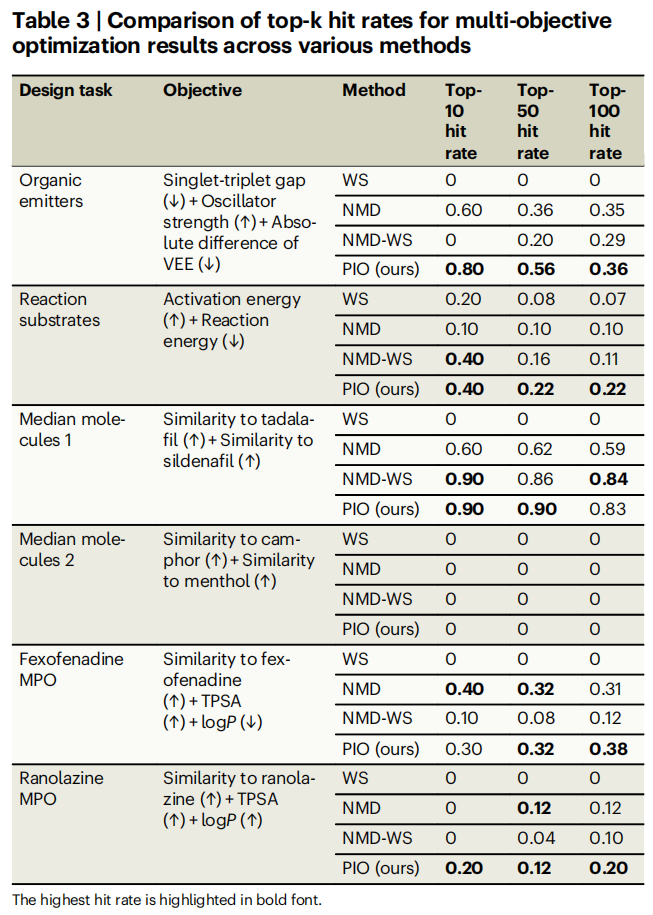
核心发现:
- • WS 方法在所有多目标任务中全部失败(命中率为 0)。 平行坐标图(Fig. 6)清楚显示,WS 倾向于过度优化某些目标(如振子强度、logP)而严重牺牲其他目标(如相似性得分)。
- • PIO 在大多数多目标任务中取得最高或并列最高的命中率,展现了通过概率乘积实现目标间自然平衡的优势。
- • NMD 和 NMD-WS 在部分任务中表现不错,但由于不考虑不确定性,仍可能在超出模型可靠范围的区域过度优化。
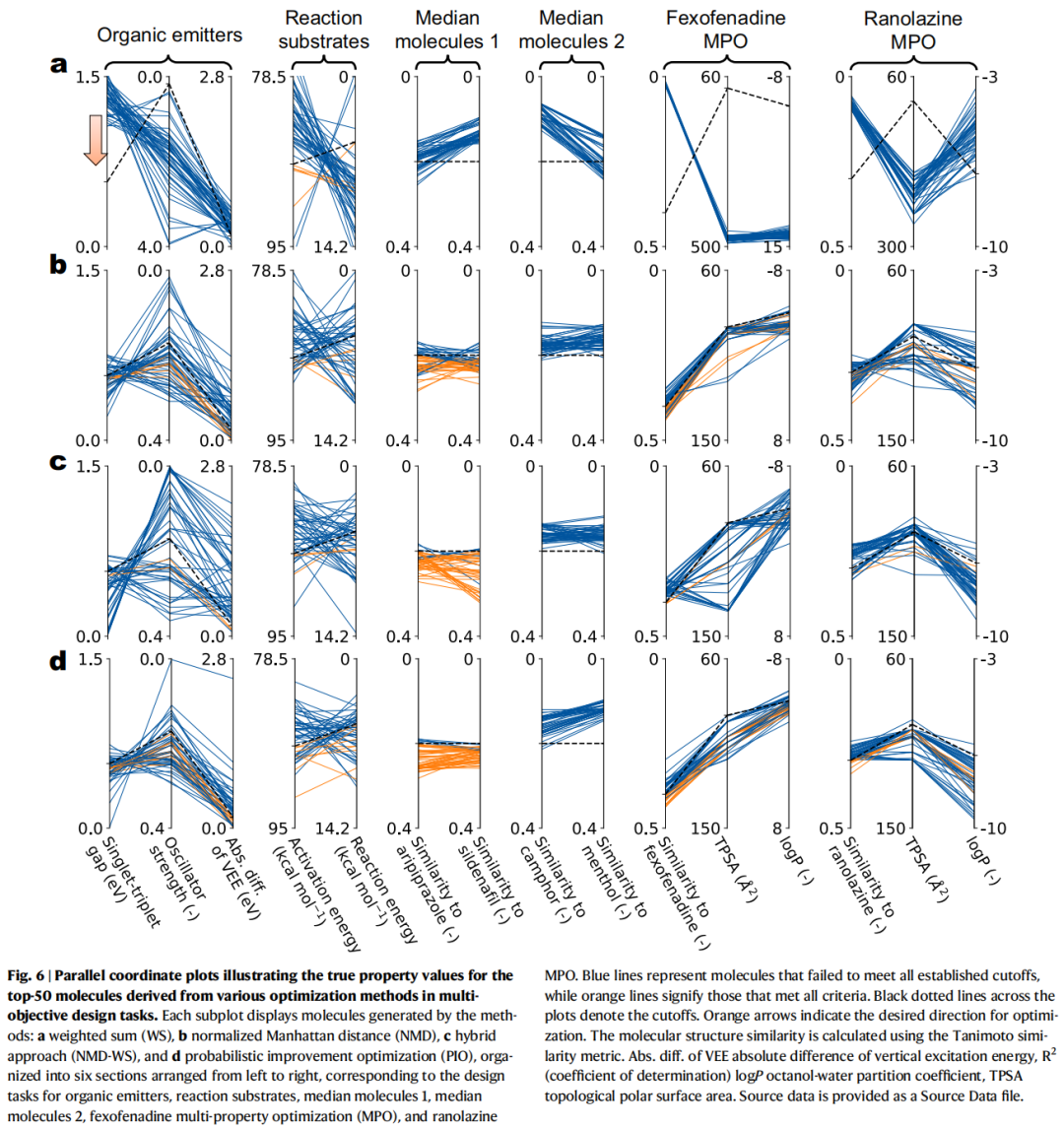
4.6 失败案例与局限性
论文坦诚讨论了三类失败场景:
(1)所有方法均失败的任务
- • 单重态-三重态间隙优化: 阈值设定过于严格,超出当前种群性能范围,导致 PIO 和 EI 的适应度得分趋近于零,失去搜索引导能力。
- • 麦雌醇相似性: 目标分子含四个稠合环的复杂多环结构,D-MPNN 难以准确表征此类复杂环结构,导致模型在广阔化学空间中的预测严重偏离真实值——且不确定性估计也不准确(过低)。
- • 中位分子 2 任务(樟脑+薄荷醇): 原始数据集中相似性得分极低(多数低于 0.1),远离目标阈值 0.2,所有方法均无法找到满足条件的分子。
(2)UQ 校准的局限
即使在测试集上校准良好的模型,在优化过程中面对高度外推区域时仍可能失效。这意味着静态的 UQ 校准不足以保证整个优化过程中的可靠性,未来需要能动态适应域偏移的 UQ 方法。
(3)阈值敏感性
PIO 和 EI 均依赖阈值设定。过于激进的阈值会使所有候选分子的适应度归零;过于宽松的阈值则削弱了 UQ 的引导作用。论文中大多数阈值设定在各任务原始数据集中 Top 分子附近的性能水平,但如何自适应地设定阈值仍是开放问题。
五、方法论贡献与启示
5.1 对 CAMD 方法论的核心启示
- 1. UQ 在开放化学空间优化中是有价值的,但需要选择正确的集成策略。 PIO 优于 DOM 和 EI 的结论,为该领域提供了明确的实践指导。
- 2. BO 中的经典策略不能机械照搬到大规模分子优化中。 EI 在 GPR + 小搜索空间中的成功不代表它适用于 GNN + 开放化学空间。搜索空间的规模和模型的不确定性行为根本不同。
- 3. 多目标优化中,概率乘积是一种优雅的目标平衡机制。 它无需手动调权,天然避免了某些目标被过度牺牲的问题。
- 4. UQ 的校准质量是方法成败的关键。 当 UQ 校准不良时,PIO 的优势减弱,这强调了发展更鲁棒的 UQ 方法的必要性。
5.2 与主动学习 / 虚拟筛选的区别
此前文献中关于"UQ 在虚拟筛选中不一定有用"的结论需要谨慎解读。虚拟筛选在固定化合物库中进行,模型面临的外推程度有限;而开放化学空间优化中,GA 不断生成全新结构,外推程度远超虚拟筛选。本文的实验清楚表明,在持续优化的开放场景下,UQ(尤其是 PIO)确实带来了实质性的性能提升。
5.3 实用性考量
PIO 方法的实用门槛很低:只需在现有 GNN 框架中添加 UQ 模块(Chemprop 已内置),设定一个合理阈值,即可将适应度函数从 DOM 切换为 PIO。代码已开源(Zenodo),可直接复用。
六、未来展望
基于论文的发现和讨论,以下几个方向值得关注:
- 1. 动态自适应 UQ: 开发能随优化过程中数据分布偏移而自动重新校准的 UQ 方法,而非依赖初始训练集上的静态校准。
- 2. 自适应阈值策略: 探索根据当前种群性能自动调整 PIO 阈值的机制,避免阈值过于激进或宽松。
- 3. 结合主动学习的闭环优化: 将 PIO 发现的高价值候选分子用于迭代更新训练集,逐步提升模型在外推区域的可靠性。
- 4. 更强的分子表征: 引入环结构指示符等额外特征增强 D-MPNN 对复杂分子(如多环体系)的建模能力。
- 5. 无分布假设的 UQ 方法: 探索保形预测(Conformal Prediction)等不依赖高斯假设的 UQ 方法与 PIO 的结合。
七、总结
这篇论文的核心贡献不在于提出了一个全新的算法,而在于对一个重要问题进行了系统性的、有深度的实证研究,并得出了具有实践指导意义的结论:
在基于 GNN 的分子优化中,概率改进优化(PIO)通过将不确定性转化为达标概率,提供了一种有界、稳定、天然平衡多目标的适应度评估策略,在广阔化学空间的探索中显著优于传统贪心方法和期望改进策略。
对于从事 CAMD、药物设计、材料发现的研究者而言,这项工作提供了一套即插即用的方法论升级路径:让你的代理模型诚实地量化自己的不确定性,然后用概率语言引导搜索方向——这比盲目信任模型预测要可靠得多。
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-04-01,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号