AlphaFold 真的学会了物理吗?——对主流蛋白质结构预测模型的系统性物理评估

AlphaFold 真的学会了物理吗?——对主流蛋白质结构预测模型的系统性物理评估

DrugIntel
发布于 2026-03-30 16:11:22
发布于 2026-03-30 16:11:22

论文信息 标题:Physics-Grounded Evaluation to Guide Accurate Biomolecular Prediction 作者:Ningyi Lyu†, Siyuan Du†, Qianzhen Shao, Zhongyue Yang, Jianpeng Ma, Daniel Herschlag 机构:复旦大学 · 斯坦福大学 · 范德堡大学 来源:bioRxiv 预印本(2026.03.25)|DOI: 10.1101/2025.06.30.662466
一、研究背景:RMSD 够用吗?
1.1 结构预测的"黄金时代"与遗留问题
AlphaFold2(2021)的出现标志着蛋白质结构预测进入了一个新纪元。凭借对数百万蛋白质的高精度预测,它在 CASP14 竞赛中以压倒性优势超越了所有传统方法,并由此催生了 AlphaFold3、ESMFold、RoseTTAFold All-Atom 等一系列后续模型。
然而,在巨大的成功光环之下,一个根本性问题始终悬而未决:
这些深度学习模型,究竟是在「记忆」结构数据,还是真正「学到」了支配生物分子行为的物理规律?
这一问题的答案,对于模型能否可靠地迁移至配体结合亲和力预测、突变效应、酶催化机制分析等下游任务至关重要。
1.2 现行评估范式的根本缺陷
当前评估生物分子预测模型最常用的指标是 RMSD(均方根偏差)——即预测结构与实验结构之间所有对应原子的平均位移距离。
RMSD 有其价值,但作者指出了一个致命的盲区:RMSD 对物理合理性完全不敏感。
以键扭转角为例说明(论文图1b):
- • 一根键从 staggered 构象(最低能量,60°)旋转至 eclipsed 构象(高能量,100°):能量升高约 3 kcal/mol(对应室温下概率降低约 160 倍),但 RMSD 仅增加约 0.4 Å;
- • 继续旋转至另一个 staggered 构象(170°):RMSD 进一步增大,但能量重新降低——预测"更像"了,其实反而"更好"了,RMSD 却持续惩罚它。
这说明 RMSD 不仅无法捕捉能量上的错误,甚至会在方向上误导模型优化。更高的 RMSD 不等于更差的物理合理性,反之亦然。
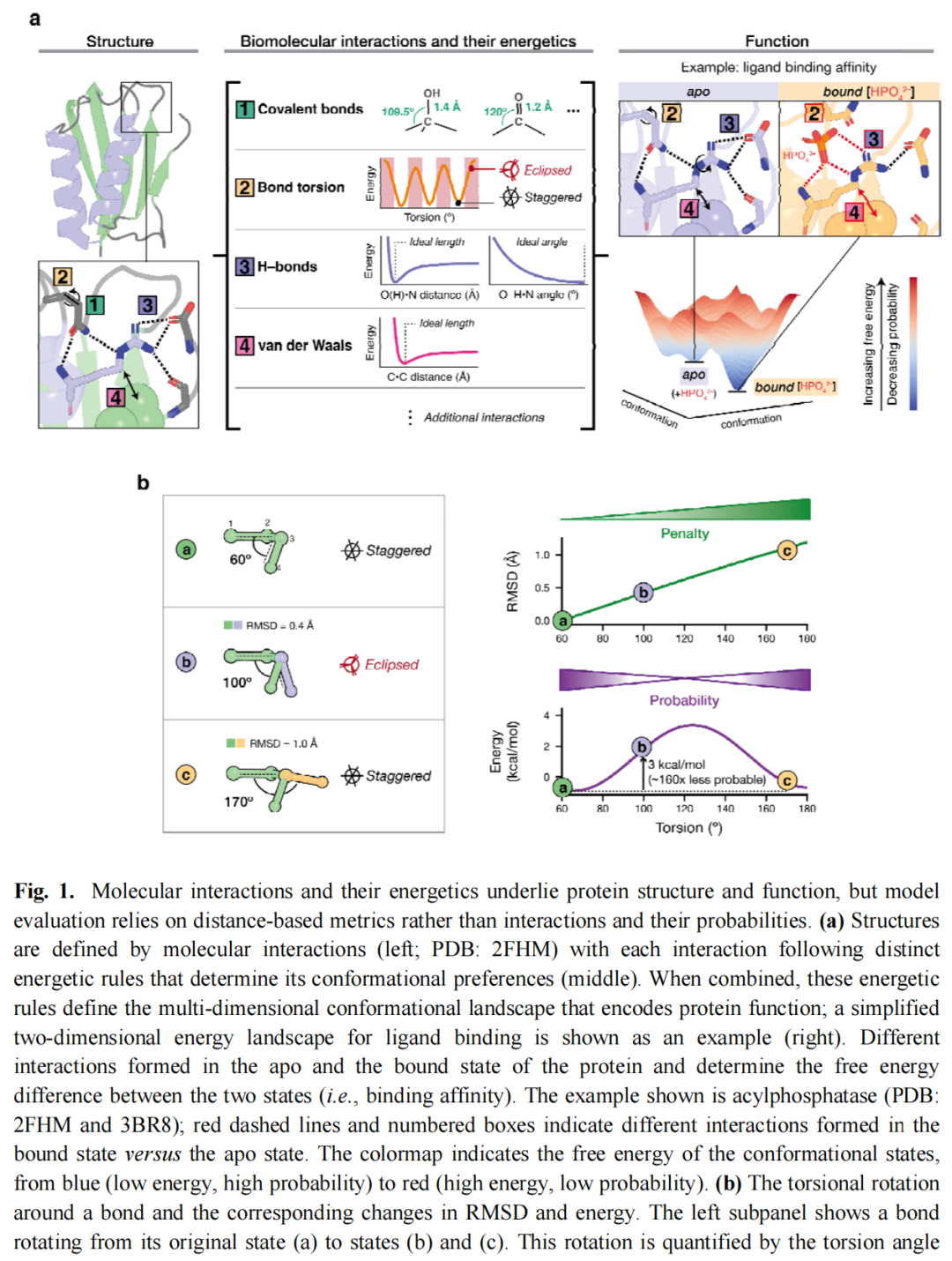

1.3 物理规律为何如此重要?
蛋白质的所有性质——折叠稳定性、配体结合亲和力、变构调控、酶催化活性——都由原子级别的相互作用网络及其能量决定。只有模型真正学到了这套物理规律,才有可能:
- 1. 在训练数据覆盖不足的区域作出可靠外推;
- 2. 准确预测对原子细节高度敏感的功能性质;
- 3. 作为下游任务(如虚拟筛选、蛋白质设计)的可靠基础。
二、研究方法:以物理量为核心的评估框架
2.1 评估指标体系
研究团队放弃了坐标距离指标,转而直接测量以下物理化学量,它们直接对应分子力学能量函数中的各项:
相互作用类型 | 评估指标 | 物理意义 |
|---|---|---|
共价键 | 键长(Å)、键角(°) | 偏离理想值意味着局部应变能 |
键扭转 | 扭转角(°)、旋转异构体状态 | staggered vs. eclipsed,决定侧链构象库 |
氢键 | 供体-受体距离(Å)、D-H···A 角度(°) | 方向性与距离决定氢键强度 |
范德华相互作用 | 原子间接触距离(Å)vs. 理想 vdW 距离 | 偏离 Lennard-Jones 极小值意味着排斥或接触缺失 |
所有分布均来源于知识库势能函数(基于 PDB 数据,温度 298 K),并通过 Boltzmann 关系转换为能量单位(kcal/mol),从而赋予分布偏差以物理可解释性。
2.2 数据集构建
- • 参考结构集:Top2018 数据集,分辨率 < 2 Å 的高质量 X 射线晶体结构,共 3939 个蛋白质链,经残基级质量过滤;
- • 预测结构:对相同序列,分别从 AF2 数据库检索 AlphaFold2 预测,以及使用 AlphaFold3 服务器和 ESMFold 生成预测;
- • 置信度:三个模型预测结构的平均 pLDDT 分别为 AF2: 96(SD=4)、AF3: 94(SD=4)、ESMFold: 89(SD=12)——总体属于高置信度预测;
- • 分析规模:共分析超过 340 万个分子相互作用;
- • 重点区域:分析聚焦于埋藏残基(相对溶剂可及性 <25%,共 41 万残基,220 万相互作用),因其构象由周围蛋白质环境的相互作用力平衡决定,且 X 射线数据建模误差最小。
2.3 基线模型设计
为区分模型性能与随机预测,研究设计了两类对照基线:
- • 基线 #1(跨旋转异构体状态):在 gauche⁻、trans、gauche⁺ 三种状态间等概率随机采样,正确率期望约 1/3;
- • 基线 #2(旋转异构体内部):在已知正确旋转异构体状态内,按 PDB 分布加权随机采样扭转角,主要采样低能 staggered 构象——此基线对 eclipsed 构象的预测偏差期望为 ~3 kcal/mol。
三、主要结果
3.1 模型学到的物理基础:成功之处
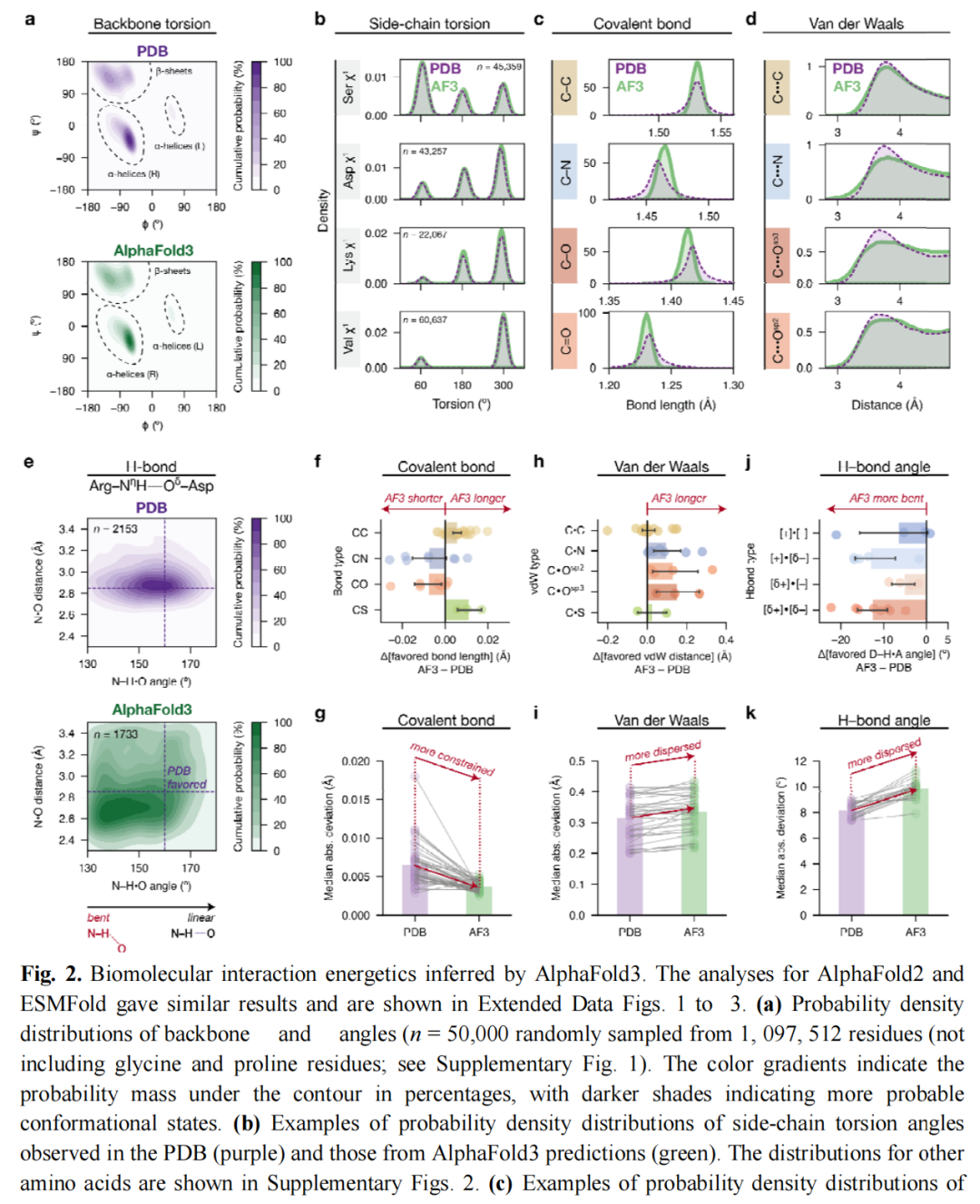
以 AlphaFold3 为主要示例,三个模型均展现出对基础物理规律的一定掌握:
骨架构象:
- • AF3 预测的骨架扭转角(φ/ψ)符合经典 Ramachandran 图分布;
- • 99% 的骨架扭转角偏差在 30° 以内;
- • 91% 的骨架肽键被正确分配到对应二级结构(α-螺旋 97%,β-折叠 96%,无规卷曲 85%)。
共价键几何:
- • AF3 正确捕捉了 C=O 键(1.22 Å)比 C–O 键(1.42 Å)短约 0.2 Å 的基本特征;
- • 侧链键对 staggered 构象表现出正确的偏好。
非共价相互作用定性规律:
- • 范德华相互作用整体符合 Lennard-Jones 形式——每种接触类型均存在偏好距离,过短距离受到惩罚;
- • 氢键存在明确的距离和角度偏好。

3.2 系统性偏差:问题所在
尽管定性规律基本正确,对分布峰值位置和宽度的精量分析揭示了显著偏差:
3.2.1 共价键几何偏差(细微但系统性)
- • 不同类型共价键的键长偏好差异约为 0.01–0.03 Å,偏差方向因原子和键类型而异;
- • 键角偏差约 1–3°;
- • AF3 的共价键分布明显窄于 PDB 分布(以 MAD 衡量),意味着模型对键长的多样性估计不足。
由于共价键几何在晶体结构精修中通常受约束,且数据量极大,这些偏差更可能源于模型本身而非训练数据的噪声。
3.2.2 非共价相互作用偏差(显著)
范德华相互作用:
- • 除 C···C 接触外,其余类型接触的偏好距离偏差均在 0.1 Å 以上;
- • AF3 的 vdW 分布宽于 PDB(更分散)。
氢键:
- • AF3 氢键采用更弯曲的几何构型(D–H···A 角度偏小),而 PDB 中及量子力学计算均显示线性氢键更受偏好;
- • 氢键分布的峰值位置偏差及分布宽度偏差尤为显著。
3.3 一对一比较:相互作用的错配
3.3.1 骨架-骨架氢键(高度准确)
几乎所有(96%)骨架·骨架氢键均在正确的原子对之间形成,这与 AF3 在蛋白质折叠预测上的卓越表现一致。
3.3.2 涉及侧链的相互作用(大量错误)
相互作用类型 | AF3 遗漏率(PDB 存在但 AF3 无) | AF3 幻觉率(AF3 有但 PDB 无) |
|---|---|---|
骨架·骨架氢键 | 4% | 4% |
骨架·侧链氢键 | 21% | 12% |
侧链·侧链氢键 | 32% | 23% |
侧链 vdW(C·S) | ~22% | ~16% |
侧链 vdW(C·O) | ~31% | ~22% |
侧链 vdW(C·N) | ~26% | ~16% |
即便是 AF3 正确识别的相互作用对,其几何参数仍存在大量偏差:
- • ~39% 的正确氢键供体-受体距离偏差 > 0.2 Å;
- • ~32% 的正确氢键弯曲程度偏差 > 20°;
- • ~32% 的正确 vdW 距离偏差 > 0.2 Å。
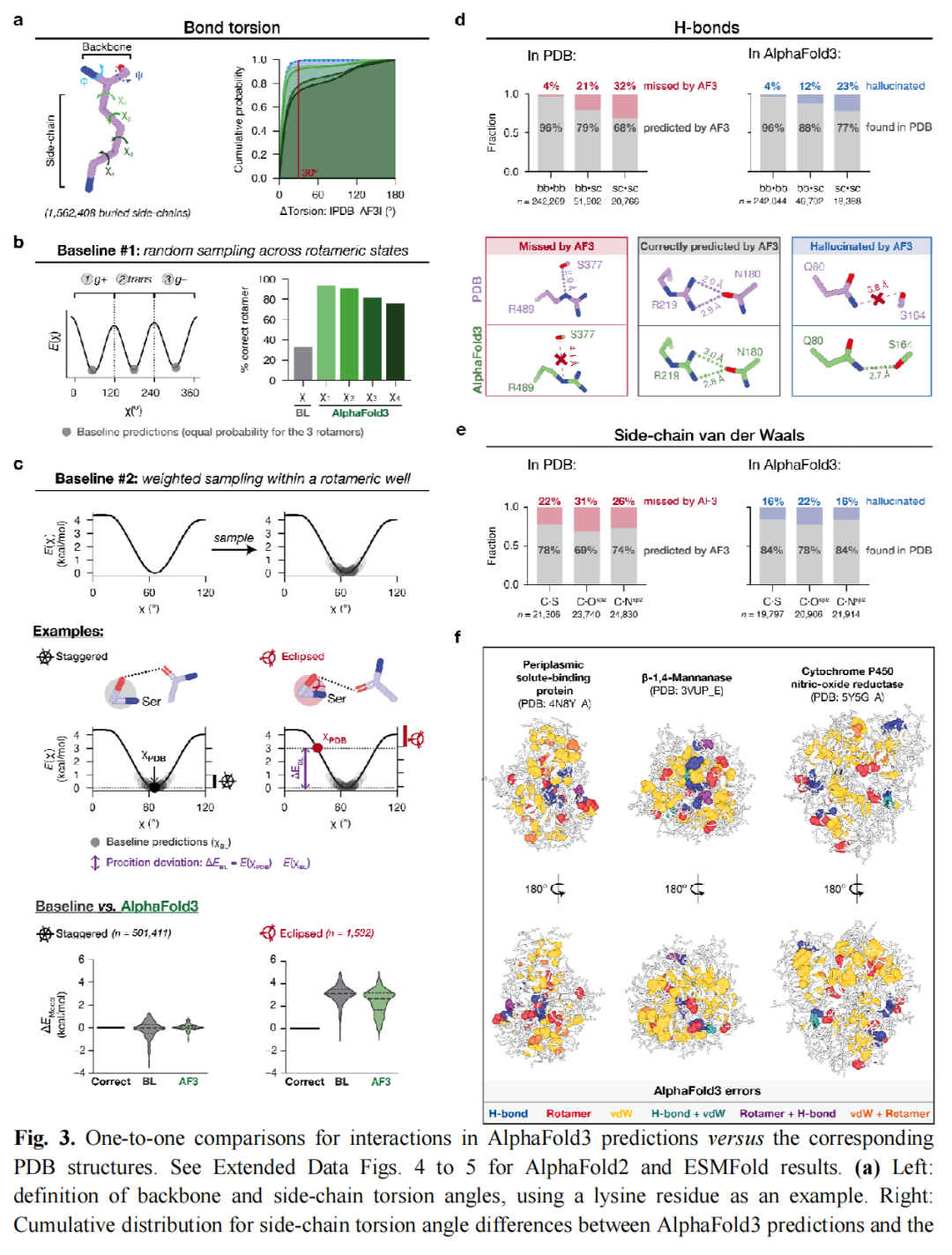

3.3.3 错误的空间分布
- • 错误预测的残基均匀分布于整个蛋白质结构中,而非集中于某些局部区域(通过最近邻距离分析验证,误差残基的空间分布与随机选取残基无显著差异);
- • 错误率与蛋白质大小成正比,与结构类型(α-富集、β-富集、混合、卷曲)无显著关联;
- • 重要警示:pLDDT 置信度分数无法识别这些错误——预测错误的相互作用所涉及残基的平均 pLDDT 高达 92(SD=8),与正确预测区域几乎相同。
3.4 侧链扭转角的精细分析
旋转异构体状态(跨能垒)的预测
AF3 对 χ₁–χ₄ 各位置的正确旋转异构体预测率分别为:
键位置 | AF3 正确率 | 基线(随机)正确率 |
|---|---|---|
χ₁ | 94% | ~33% |
χ₂ | 92% | ~33% |
χ₃ | 78% | ~33% |
χ₄ | 73% | ~33% |
随距骨架越远,准确率下降,可能因为:(1) 更大的构象空间;(2) 更多的相互作用影响;(3) 数据稀疏性。
旋转异构体内部(精细能量平衡)的预测
在正确旋转异构体状态内,模型能否捕捉 eclipsed 等高能构象?
- • 对 PDB 中处于 2.5–4.0 kcal/mol 高能 eclipsed 状态的键(n=1532),AF3 平均预测偏差为 ΔE = 2.4 kcal/mol(SD=1.1);
- • 基线模型的期望偏差为 3.1 kcal/mol(SD=0.7);
- • 仅 6% 的 eclipsed 键被 AF3 在 0.5 kcal/mol 以内正确预测。
这表明模型对局部能量平衡的精细捕捉能力仍严重不足——而这正是预测侧链非共价相互作用的核心所在。
3.5 构象系综:AI 世界的"过度确定性"
利用多温度(MT)X 射线晶体学数据(9 个蛋白质,分辨率 1.0–2.2 Å,200 K 以上采集),研究团队评估了 AF3 多次随机种子采样的构象多样性。
实验数据显示的多构象态:
- • 在 1363 个埋藏侧链键中,136 个键(270 个构象体)明确占据多于一种构象状态。
AF3 随机采样的结果:
- • 136 个多构象键中,96 个(71%) 在 AF3 采样中仅产生单一构象,分布极窄;
- • 仅 40 个(29%)被预测为多构象,其中 38 个在方向上与 MT 模型一致(高精度),仅 2 个为幻觉。
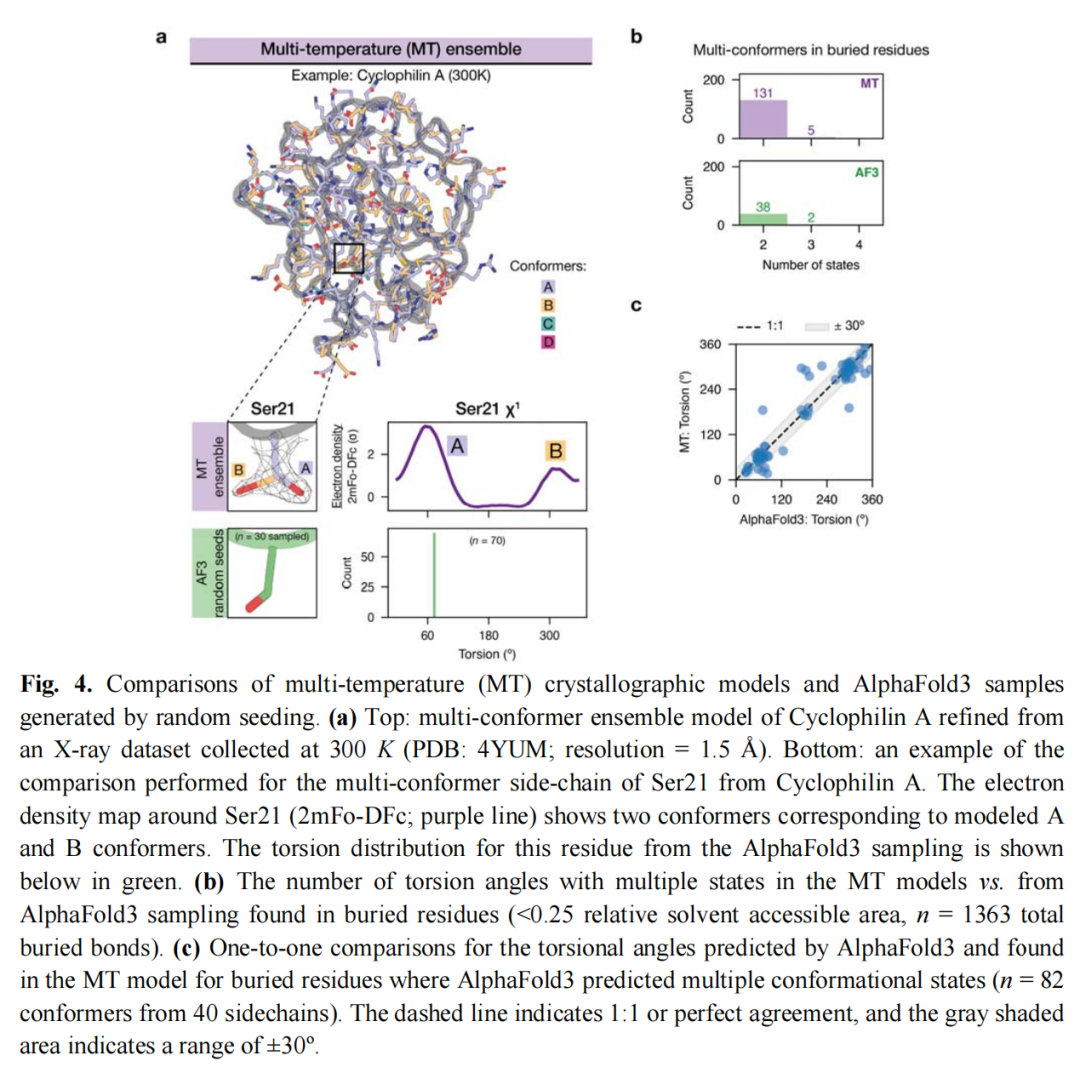
结论: AF3 的内部概率景观被严重压缩,即使使用随机种子多次采样,输出也几乎是确定性的。这对于需要捕捉构象灵活性的功能预测任务是重大局限。
四、模型间对比:各有千秋,共同踩坑
4.1 ESMFold:最大的非共价相互作用缺陷
ESMFold 在骨架构象和扭转角偏好方面表现与 AF2/AF3 相当,但在非共价相互作用上表现最差:
- • 侧链·侧链氢键遗漏率高达 ~58%;
- • vdW 相互作用遗漏率 ~33–43%;
- • 其非共价相互作用分布极度弥散,偏好峰值偏差最大。
这与既往研究中 ESMFold 整体结构精度较低的结论一致,但本研究进一步定位了其主要错误来源——非共价成对相互作用。
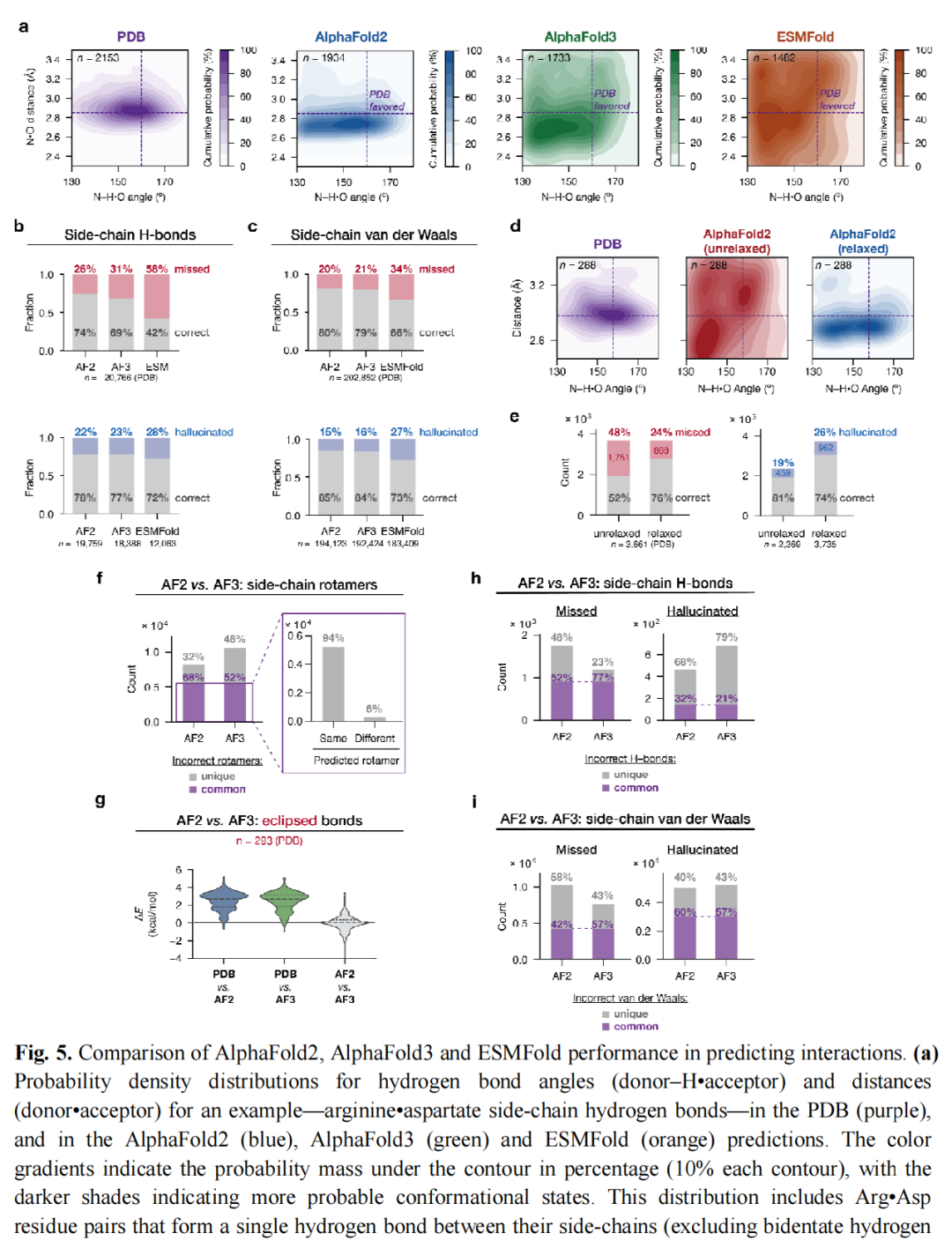

4.2 AlphaFold2 vs. AlphaFold3:架构不同,错误相似
AF2 在侧链·侧链氢键方面略好于 AF3(26% vs. 31%),这主要归功于其 AMBER 力场弛豫后处理步骤。
力场弛豫的效果与局限:
指标 | AF2 未弛豫 | AF2 弛豫后 |
|---|---|---|
侧链·侧链氢键遗漏率 | 48% | 24% |
侧链·侧链氢键幻觉率 | 19% | 26%(略有上升) |
vdW 遗漏率改善 | — | ~8% |
对 AF3 进行相同弛豫处理,结果与 AF2 相似(从 32% 降至 22%),弛豫后两者错误水平接近。
共同错误的惊人发现:
AF2 和 AF3 的共同错误覆盖令人警惕:
错误类型 | 共同错误比例 |
|---|---|
旋转异构体错误 | ~68%(5551/~9000 总错误中) |
共同错误中,两者预测了相同的错误旋转体 | 94% |
PDB 氢键被两者同时遗漏 | ~77% |
两者同时幻觉的氢键 | ~21% |
PDB vdW 被两者同时遗漏 | ~57% |
两者同时幻觉的 vdW | ~57% |
力场弛豫后,剩余错误仍大量重叠(见论文图S13)。
这一发现提示两种可能机制:(1) AF3 使用了 AF2-Multimer 的预测结果扩充训练集,引入了共同偏差;(2) 某些特定位点的能量平衡本身极难从 PDB 数据中学习。
五、机制讨论:为何会出现这些偏差?
5.1 训练目标的根本局限
当前模型的训练逻辑主要以坐标匹配为目标(如均方误差损失函数),而非直接优化物理相互作用的准确性。这导致:
- • 对旋转异构体的整体状态(需较大空间移动,RMSD 惩罚明显)有较好的学习压力;
- • 对旋转异构体内部的精细能量平衡(RMSD 变化极小但能量变化显著)几乎没有梯度信号。
这解释了为何模型在"大方向"上(正确旋转异构体状态)表现尚可,而在"精细能量"层面(eclipsed vs. staggered 的区分,氢键方向,vdW 距离精度)严重失准。
5.2 训练数据的质量问题
- • PDB 中低分辨率结构存在系统性坐标误差,尤其是侧链位置;
- • 冷冻晶体学条件下,蛋白质构象多样性被严重低估,无法提供系综信息;
- • 训练集中 apo 与配体结合态结构混合,可能引入对结合口袋区域的偏差(虽本研究验证影响有限)。
5.3 AF3 扩散架构的特殊性
AlphaFold3 采用了基于扩散的全原子生成架构,可以不受预设约束地自由生成坐标。这带来:
- • 优势:理论上更能捕捉非共价相互作用的细节(共价键几何不被硬约束);
- • 现实:内部概率景观的"收缩"导致构象采样多样性极低,系综生成能力受限。
六、对下游应用的影响
这些发现对以下实际应用具有直接警示意义(见论文补充表1):
下游任务 | 受影响的主要因素 | 建议谨慎程度 |
|---|---|---|
配体对接与虚拟筛选 | 结合口袋侧链相互作用错误 | ⚠️ 高度谨慎 |
突变效应(ΔΔG)预测 | 突变位点局部相互作用网络失真 | ⚠️ 高度谨慎 |
蛋白质热稳定性预测 | 疏水核心 vdW 接触错误 | ⚠️ 中度谨慎 |
蛋白质-蛋白质相互作用界面 | 界面侧链氢键和极性接触错误 | ⚠️ 中度谨慎 |
蛋白质折叠拓扑预测 | 骨架构象整体可靠 | ✅ 相对可信 |
替代构象与变构研究 | 系综多样性严重低估 | ⚠️ 高度谨慎 |
七、改进方向与未来展望
7.1 训练数据层面
- • 多温度 X 射线晶体学数据:提供构象系综信息,帮助模型学习能量景观而非单一构象;
- • 扰动晶体学(电场、压力晶体学):提供蛋白质对物理扰动的动力学响应数据;
- • 高分辨率冷冻电镜数据:在接近生理条件下提供系综信息;
- • 严格分离 apo/bound 训练数据:减少混合状态引入的系统性偏差。
7.2 训练目标层面
- • 在损失函数中显式加入物理能量项(键长、键角、扭转能、氢键能、vdW 能),并增大其权重;
- • 借鉴小分子生成领域的成功经验——物理引导的基准测试(如 PoseBusters)极大推动了分子生成模型的改进;
- • 引入知识库势能函数(knowledge-based potential)作为辅助训练目标。
7.3 后处理层面
- • 力场弛豫(如 AMBER)是有效的补充手段,但需对弛豫结果本身进行物理评估;
- • 分子动力学模拟可进一步修正侧链构象,但也需物理框架下的严格验证。
7.4 评估框架的推广
本研究提出的框架天然可扩展至:
- • 核酸结构预测(磷酸骨架扭转、碱基堆叠、Watson-Crick 氢键);
- • 小分子配体的结合姿态评估(替代 RMSD 为主的 docking 评分);
- • 蛋白质-核酸、蛋白质-配体复合物的相互作用界面;
- • 所有输出原子坐标的生物分子预测模型(RoseTTAFold、ESMFold、Chai-1 等)。
八、方法论的历史意义
这一工作与小分子生成领域的发展脉络有深刻相似之处。早期分子生成模型(VAE、GAN 等)产生了大量含有非法价态和高应变几何的分子,被药物化学家视为"无用"。正是一系列以物理为核心的基准测试(Fréchet ChemNet Distance、GuacaMol、PoseBusters 等)的出现,推动了后续模型几乎完全消除价态错误、大幅减少高能构象,从而真正走向实用。
蛋白质结构预测领域正面临同样的转折点:从以坐标精度为中心的评估范式,转向以物理合理性和功能可预测性为中心的评估范式。
九、总结
维度 | 核心结论 |
|---|---|
已学到的物理 | 骨架构象(Ramachandran 分布)、侧链旋转异构体状态(94% χ₁ 正确)、共价键长基本差异、vdW 定性规律 |
尚未学到的物理 | 精细能量平衡(eclipsed 构象预测仅 6% 正确)、氢键方向性、侧链 vdW 精确距离、构象系综多样性 |
错误规模 | 侧链·侧链氢键:AF2 26%,AF3 32%,ESM ~58%;vdW 类似量级 |
错误分布 | 全结构随机分布,pLDDT 无法识别 |
模型间关联 | AF2/AF3 共享 68% 旋转异构体错误,77% 共同遗漏相同氢键 |
力场弛豫效果 | 将 AF2 侧链氢键错误从 48% 降至 24%,但仍残留 >20% 错误,且引入新幻觉 |
系综生成 | 71% 的多构象侧链被 AF3 预测为单构象,内部概率景观过度收缩 |
应用启示 | 配体对接、突变效应等需谨慎;折叠拓扑预测相对可靠 |
改进方向 | 物理约束训练目标 + 系综训练数据 + 逐步评估驱动的开发范式 |
本文参与 腾讯云自媒体同步曝光计划,分享自微信公众号。
原始发表:2026-03-27,如有侵权请联系 cloudcommunity@tencent.com 删除
评论
登录后参与评论
推荐阅读
目录
腾讯云开发者

Copyright © 2013 - 2026 Tencent Cloud. All Rights Reserved. 腾讯云 版权所有
深圳市腾讯计算机系统有限公司 ICP备案/许可证号:粤B2-20090059 ![]() 粤公网安备44030502008569号
粤公网安备44030502008569号
腾讯云计算(北京)有限责任公司 京ICP证150476号 | 京ICP备11018762号